p
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington D. C. 20549
FORM
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
or
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from
Commission file number
(Exact name of registrant as specified in its charter)
|
|
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
(Address of principal executive offices) (Zip Code)
(
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
The |
Securities registered pursuant to Section 12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer as defined in Rule 405 of the Securities Act. ☐ Yes ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ☐ Yes ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer”, “accelerated filer”, “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer |
|
☐ |
|
Accelerated filer |
☐ |
|
☒ |
|
Smaller reporting company |
||
Emerging growth company |
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
Indicate by check mark whether the registrant is a shell company (as defined by Rule 12b-2 of the Act)
As of December 31, 2021, the aggregate market value of the issued and outstanding common stock held by non-affiliates of the registrant, based upon the closing price of our common stock of $0.51 was approximately $
Number of shares of common stock outstanding as of September 26, 2022 was
DOCUMENTS INCORPORATED BY REFERENCE –
Auditor Name: Marcum LLP Auditor Location: San Francisco, CA Auditor Firm ID:
FORM 10-K
FOR THE FISCAL YEAR ENDED JUNE 30, 2022
TABLE OF CONTENTS
i
PART I
Item 1. Business.
Background
Kintara Therapeutics, Inc. is a clinical stage, biopharmaceutical company focused on the development and commercialization of new cancer therapies.
On June 10, 2020, we entered into an Agreement and Plan of Merger and Reorganization (the “Merger Agreement”) dated as of June 9, 2020, by and among Adgero Acquisition Corp., our wholly-owned subsidiary incorporated in the State of Delaware (“Merger Sub”), and Adgero Biopharmaceuticals Holdings, Inc., a Delaware corporation (“Adgero”). On August 19, 2020, upon the terms and subject to the conditions set forth in the Merger Agreement, Merger Sub merged with and into Adgero (the “Merger”), the separate corporate existence of Merger Sub ceased, and Adgero continued its existence under Delaware law as the surviving corporation in the Merger and became our direct, wholly-owned subsidiary.
Following the completion of the Merger, we changed our name from DelMar Pharmaceuticals, Inc. to Kintara Therapeutics, Inc. and began trading on The Nasdaq Capital Market LLC under the symbol “KTRA”.
We are the parent company of Del Mar Pharmaceuticals (B.C.) Ltd. ("Del Mar (BC)"), a British Columbia, Canada corporation, and Adgero. We are also the parent company to 0959454 B.C. Ltd. ("Callco") and 0959456 B.C. Ltd. ("Exchangeco") which are British Columbia, Canada corporations. Callco and Exchangeco were formed to facilitate the reverse acquisition that occurred in 2013. In addition, we are the parent company to Adgero Biopharmaceuticals Inc. ("Adgero Bio").
References to “we”, “us”, and “our”, refer to Kintara and our wholly-owned subsidiaries, Del Mar (BC), Adgero, Adgero Bio, Callco, and Exchangeco.
We are dedicated to the development of novel cancer therapies for patients with unmet medical needs. Our mission is to benefit patients by developing and commercializing anti-cancer therapies for patients whose solid tumors exhibit features that make them resistant to, or unlikely to respond to, currently available therapies, with particular focus on orphan cancer indications.
Our two lead candidates are VAL-083, a novel, validated, DNA-targeting agent, for the treatment of drug-resistant solid tumors such as glioblastoma multiforme (“GBM”) and potentially other solid tumors, including ovarian cancer, non-small cell lung cancer (“NSCLC”), and diffuse intrinsic pontine glioma (“DIPG”) and REM-001, a late-stage photodynamic therapy (“PDT”) for the treatment of cutaneous metastatic breast cancer (“CMBC”), basal cell carcinoma nevus syndrome (“BCCNS"), and access graft failure in hemodialysis patients. PDT is a treatment that uses light sensitive compounds, or photosensitizers, that, when exposed to specific wavelengths of light, act as a catalyst to produce a form of oxygen that induces local tumor cell death.
Recent Highlights
2
Targeted Clinical Milestones
(Calendar Quarters)
Below are our planned, or expected, milestones for the respective time periods noted:
Around the end of Q3 2022
Around the end of Q3 2023
Around the end of Q4 2023
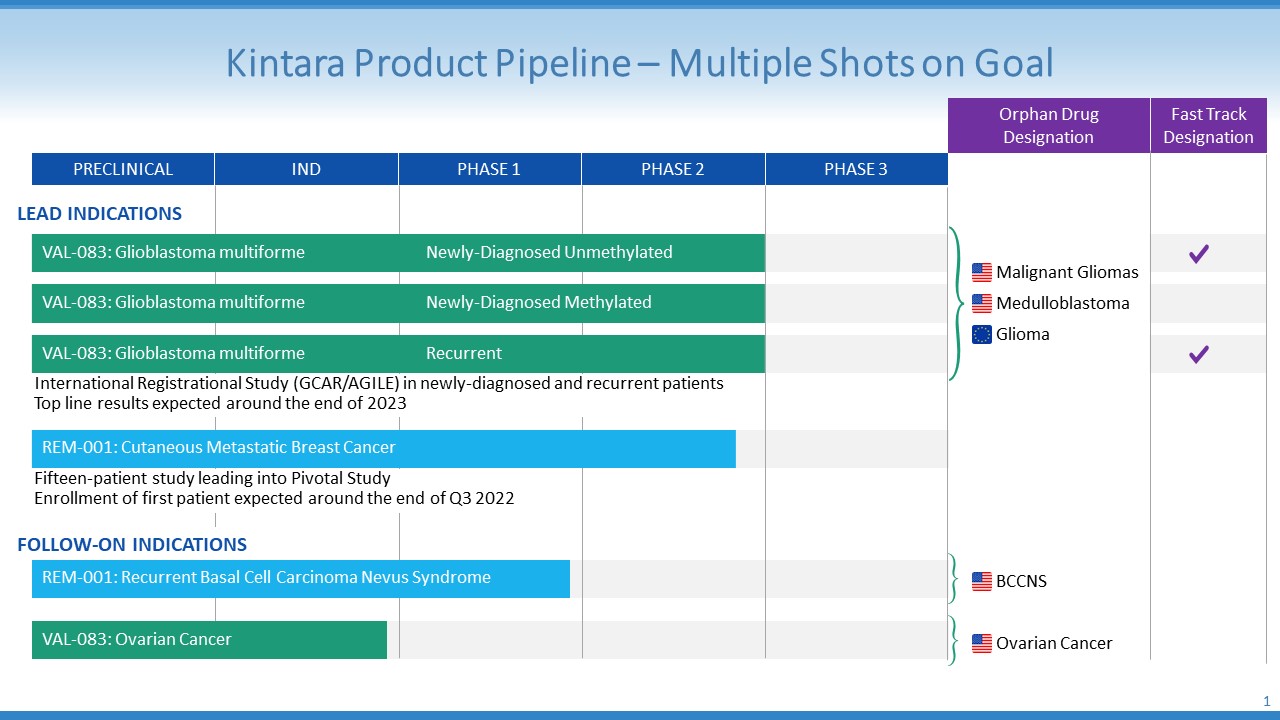
Product Pipeline
3
VAL-083
Background
VAL-083 is a first-in-class, small-molecule, DNA-targeting chemotherapeutic that has demonstrated activity against a range of tumor types in prior Phase 1 and Phase 2 clinical studies sponsored by the US National Cancer Institute (“NCI”). “First-in-class” means that VAL-083 embodies a unique molecular structure which is not an analogue, or derivative, of any approved product, or product under development, for the treatment of cancer. As part of our business strategy, we leverage and build upon these prior NCI investments and data from more than 40 NCI Phase 1 and Phase 2 clinical studies, which includes an estimated 1,200 patient safety database.
In GBM, we are part of the GBM AGILE Study which is a registrational Phase 2/3 clinical study for GBM. The study is a revolutionary, patient centered, adaptive platform study for registration evaluating multiple therapies for patients with newly-diagnosed and recurrent GBM. Patients in the GBM AGILE Study are tested for their O6-methyl guanine methyltransferase ("MGMT") methylation status prior to enrollment. VAL-083 is being evaluated in all three GBM patient subtypes in this study: newly-diagnosed methylated MGMT; newly-diagnosed unmethylated MGMT; and recurrent.
We have also completed two open-label, biomarker-driven, Phase 2 studies in MGMT-unmethylated GBM. MGMT is a DNA-repair enzyme that is associated with resistance to temozolomide ("TMZ" or Temodar®), the current standard-of-care chemotherapy used in the treatment of GBM. More than 60% of GBM patients have MGMT-unmethylated tumors and exhibit a high expression of MGMT which is correlated with TMZ treatment failure and poor patient outcomes as indicated in the current National Comprehensive Cancer Network (“NCCN”) guidelines for GBM treatment. Our research demonstrates that VAL-083’s anti-tumor activity is independent of MGMT expression. In our completed Phase 2 studies we used MGMT as a biomarker to identify patients for treatment with VAL-083 in three distinct GBM patient populations.
In addition, we have undertaken research in ovarian cancer. Ovarian cancer is the fifth most common cancer in women and is the leading cause of death among women diagnosed with gynecological malignancies. We are in the process of evaluating the best path forward in ovarian cancer including the potential combination of VAL-083 with PARP inhibitors. The FDA granted orphan drug designation for the use of VAL-083 in the treatment of ovarian cancer.
We have a broad patent portfolio to protect our intellectual property. Our patents and patent applications claim methods of use of VAL-083 and related compounds, synthetic methods, and quality controls for the manufacturing process of VAL-083. We believe that our portfolio of intellectual property rights provides a defensible market position for the commercialization of VAL-083. In addition, VAL-083 has been designated by the FDA as an orphan drug under the Orphan Drug Act and the European Medicines Agency (“EMA”) for the treatment of gliomas, including GBM. The FDA has also granted Orphan Drug description to VAL-083 for the treatment of medulloblastoma and ovarian cancer.
Our corporate strategy is to advance VAL-083 and REM-001 on an indication-by-indication basis, and then to consider out-licensing when a corporate development program has matured enough to warrant proper licensing valuations. In addition to VAL-083’s applicability to multiple solid tumor indications, we are also constantly evaluating licensing or acquiring additional product candidates, in order to establish a product pipeline and to position us for long-term sustainability and growth of shareholder value. We believe the experience of our clinical development team will position us to efficiently develop possible drug candidates that we may acquire, or license, in the future.
We intend to continue to evaluate options for our strategic direction. These options may include raising additional capital, the acquisition of another company and/or complementary assets, our sale, or another type of strategic partnership.
MGMT-unmethylated GBM
GBM is the most common and the most lethal form of glioma. Approximately 30,000 new cases of GBM are diagnosed per year in the United States and Europe combined. Within the GBM patient population, over 60% of patients are unmethylated with respect to their MGMT status.
Measurement of MGMT methylation status has become routine in clinical practice as a biomarker that correlates with resistance to the standard-of-care chemotherapy with TMZ and patient outcomes in GBM. Over 60% of GBM patients’ tumors are characterized as “MGMT-unmethylated” and exhibit a high expression of MGMT, a naturally occurring DNA-repair enzyme, the activity of which nullifies the chemotherapeutic activity of TMZ. The lack of specific therapies for MGMT-unmethylated GBM is a significant unmet medical need. Current NCCN guidelines state that the treatment benefit of TMZ is likely to be lower in GBM patients with an unmethylated MGMT promoter.
4
We have demonstrated that VAL-083’s anti-tumor mechanism is active independent from the MGMT status in vitro. We believe this distinct mechanism of action suggests the potential of VAL-083 as a replacement for the current standard-of-care chemotherapy, TMZ, in both MGMT methylated and MGMT-unmethylated GBM. We have utilized MGMT-methylation status to identify GBM patients who are unlikely to respond to TMZ and have included only MGMT-unmethylated patients in our current Phase 2 clinical studies of VAL-083. We have recently received approval to treat newly-diagnosed methylated, newly-diagnosed unmethylated, and recurrent GBM patients as part of our treatment arm in the GCAR GBM AGILE study.
We believe that our research highlights the opportunity for VAL-083 as a potential new standard-of-care in the treatment of both MGMT methylated and MGMT-unmethylated GBM.
VAL-083 Clinical Studies
GBM AGILE Study
On October 21, 2020, we announced we had entered into a definitive agreement with GCAR and on January 13, 2021, we announced the initiation of patient recruitment for the VAL-083 study arm of the GBM AGILE Study. VAL-083 is currently being evaluated in all three GBM patient subtypes in the GBM AGILE Study: newly-diagnosed methylated MGMT; newly-diagnosed unmethylated MGMT; and recurrent. The GBM AGILE Study employs a cost-efficient, adaptive study design with a stage 1 (Phase 2) learning and adapting phase and a stage 2 (Phase 3) expansion and confirmation phase. The GBM AGILE Study is currently enrolling patients in our arm of the study at 39 clinical sites in the United States, four in Canada, and two in Europe. GCAR plans to enroll 150-200 patients in the Kintara arm of the study at over 40 sites in the U.S. and Canada with potential to increase this total to 65 clinical study centers worldwide. The GBM AGILE Study, which was designed by GCAR with input from the FDA, calls for companies participating in this platform study to only disclose data at the end of the study to protect the integrity of the trial arm data. Therefore, we expect to announce topline data from the GBM AGILE Study around the end of calendar 2023.
GCAR has previously announced that the GBM AGILE Study has screened over 1,000 patients and that enrollment rates for the study are 3 to 4 times greater than traditional GBM studies, with active sites averaging 0.75 to 1 patient per site per month. As a result of the accelerated enrollment rate, we expect to announce topline data from our arm of the study around the end of calendar year 2023.
The GBM AGILE Study is an international, innovative platform study designed to more rapidly identify and confirm effective therapies for patients with glioblastoma through response adaptive randomization and a seamless Phase 2/3 design. The study, conceived by over 130 key opinion leaders, is conducted under a master protocol, allowing multiple therapies or combinations of therapies from different pharmaceutical partners to be evaluated simultaneously. With its innovative design and efficient operational infrastructure, we believe data from the GBM AGILE Study can be used as the foundation for a New Drug Application (“NDA”) and biologics license application submissions and registrations to the FDA and other health authorities.
GCAR is a 501(c)(3) nonprofit organization uniting physicians, clinical researchers, advocacy and philanthropic organizations, biopharma, health authorities, and other key stakeholders in healthcare to expedite the discovery and development of treatments for patients with rare and deadly diseases by serving as a sponsor of innovative and complex studies including master protocols and platform studies. GCAR is the sponsor of GBM AGILE. Key strategic partners for the GBM AGILE study effort include the National Brain Tumor Society (“NBTS”), National Foundation for Cancer Research, and Asian Fund for Cancer Research.
Phase 2 Study in Newly-Diagnosed MGMT-unmethylated GBM
In September 2017, we initiated a single arm, biomarker driven, open-label Phase 2 study in newly-diagnosed MGMT-unmethylated GBM patients at Sun Yat-sen University Cancer Center (“SYSUCC”) in Guangzhou, China. The study was conducted under our collaboration agreement with Guangxi Wuzhou Pharmaceutical Company.
In this Phase 2 study, VAL-083 was combined with radiotherapy as a potential replacement for standard-of-care chemoradiation with TMZ in patients with MGMT-unmethylated GBM. The goals of the study were to confirm the safety of the three-day VAL-083 dosing regimen in combination with radiotherapy and to investigate efficacy outcomes of the combination of VAL-083 and radiotherapy in MGMT-unmethylated GBM patients.
We have completed enrollment of this study with a total of 29 newly-diagnosed, MGMT-unmethylated GBM patients and we have also completed treatment of the patients on this study. The efficacy endpoints of the study include tumor response, as assessed by the Response Assessment in NeuroOncology (“RANO”), and progression-free survival (“PFS”), progression-free survival at six months (“PFS6”), and overall survival (“OS”), compared to historical results in the target population. The study was conducted in two parts: (1) Dose-confirmation: VAL-083 in cohorts (20, 30 and 40 mg/m2/day IV daily x 3 every 21 days) to assess safety and activity when administered concurrently with x-ray therapy (“XRT”) to confirm the maximum tolerated dose (“MTD”), and (2) Expansion:
5
VAL-083 was studied in 20 additional patients at the target dose, as determined by the dose-confirmation part of the study, administered concurrently with XRT. Assessments of safety and tolerability were used to support further clinical development of VAL-083 in combination with radiotherapy. Pharmacokinetic assessments of VAL-083 in plasma and cerebral spinal fluid (“CSF”) were used to correlate drug exposure in the central nervous system with patient outcomes.
Dose-confirming cohorts studying 20, 30, and 40 mg/m2/day x three every 21 days have been completed. Based on the dose confirmation phase of the study, we have selected 30 mg/m2/day for combination with irradiation for the treatment of newly-diagnosed MGMT-unmethylated GBM patients. This study was fully enrolled at 29 patients.
On April 10, 2021, at the virtual AACR Annual Meeting, we provided top-line results on patient data as follows:
While this was not a head-to-head study, this PFS and mOS data compares favorably to historical TMZ control data of 5.0 months and 6.9 months PFS and 12.7 months and 16.0 months mOS as indicated by published data from Hegi et al. (2005 - New England Journal of Medicine), Tanguturi et al. (2017 – NeuroOncology), and Alnahhas et al. (2020 - Neurooncol Adv), respectively.
Multiple treatment cycles of VAL-083 at the 30 mg/m2/day dose in combination with standard radiation treatment (2 Gray/day, 5 days/week) was shown to be generally safe and well-tolerated.
Phase 2 Study in Recurrent GBM and Newly-Diagnosed MGMT-unmethylated GBM
Recurrent Study Arm
In February 2017, we initiated a biomarker driven, open-label, single-arm Phase 2 study in collaboration with the University of Texas MD Anderson Cancer Center ("MD Anderson") for recurrent GBM patients. This biomarker-driven study (testing for MGMT methylation status) has been completed. The study enrolled a total of 89 patients with 35 patients (35 efficacy evaluable) initially receiving a dose of VAL-083 at 40 mg/m2/day, and 54 patients (48 efficacy evaluable) initially receiving the treatment dose of 30 mg/m2/day on days 1, 2 and 3 of a 21-day cycle. This 30 mg dose corresponds to the dose being studied in the currently enrolling VAL-083 study arm of the GBM AGILE study.
The study was designed to determine the potential of VAL-083 treatment to improve overall survival in GBM patients whose tumors have recurred following treatment with TMZ. These patients will not have been treated previously with Avastin®.
On November 18, 2021, at the Society for Neuro-Oncology (“SNO”) Annual Meeting we reported top-line patient data as follows:
While this was not a head-to-head study, historically, lomustine, which is the most commonly used chemotherapy for these patients, has demonstrated mOS of 7.2 months as indicated by published data from Wick et al. (2017 – New England Journal of Medicine).
All patients have completed treatment. A detailed description of this study can be found at clinicatrials.gov, Identifier Number: NCT02717962.
6
Newly-Diagnosed Adjuvant Study Arm
On July 24, 2019, we announced the enrollment of the first patient in the newly-diagnosed adjuvant arm of the Phase 2 study being conducted at MD Anderson. The newly-diagnosed adjuvant arm was originally planned for 24 patients, but based on encouraging outcomes, we increased the newly-diagnosed adjuvant arm enrollment from the originally planned 24 patients to include up to 12 additional patients. These patients will have had initial cycles of TMZ concomitant with radiation but will not have yet started subsequent cycles of TMZ (i.e., maintenance stage TMZ patients).
On November 18, 2021, at the SNO Annual Meeting we reported top-line patient data as follows:
While this was not a head-to-head study, this PFS and mOS data compares favorably to historical TMZ control data of 5.0 months and 6.9 months PFS and 12.7 months and 16.0 months mOS as indicated by published data from Hegi et al. (2005 - New England Journal of Medicine), Tanguturi et al. (2017 – NeuroOncology), and Alnahhas et al. (2020 - Neurooncol Adv), respectively
Based on published data from our MD Anderson and SYSUCC clinical studies, we believe there is a significant opportunity to treat GBM patients in the pre-TMZ maintenance stage (i.e., adjuvant).
All patients have completed treatment. A detailed description of this study can be found at clinicaltrials.gov, Identifier Number: NCT02717962.
Safety Across Studies
Consistent with prior studies, myelosuppression was the most common adverse event with VAL-083 in both the recurrent GBM and adjuvant treatment setting at MD Anderson. In the 30 mg/m2/day starting dose cohort (the dose being studied in the GBM AGILE Study) five subjects have experienced a serious adverse event (“SAE”) possibly related to VAL-083 in the recurrent group and one patient experienced a possible drug-related SAE in the newly-diagnosed adjuvant group.
In the newly-diagnosed first-line study completed at SYSUCC, three subjects experienced an SAE possibly related to VAL-083. Multiple treatment cycles of VAL-083 at the 30 mg/m2/day dose in combination with standard radiation treatment (2 Gray/day, 5 days/week) were shown to be generally safe and well-tolerated.
VAL-083 Fast Track Designation
The FDA has granted us FTD for VAL-083 in recurrent and newly-diagnosed unmethylated GBM.
The FTD is designed to expedite the review of drugs that show promise in treating life-threatening diseases and address unmet medical needs, with the goal of getting new treatments to patients earlier. FTD provides sponsors with an opportunity for increased frequency for communication with the FDA to ensure an optimal development plan and to collect appropriate data needed to support drug approval. Additional benefits of the FTD may include an Accelerated Approval, a Priority Review, and a Rolling Review. Accelerated Approval is granted to drugs that demonstrate an effect on a surrogate, or intermediate endpoints, reasonably likely to predict clinical benefit. Priority Review shortens the FDA review process for a new drug from ten months to six months and is appropriate for drugs that demonstrate significant improvements in both safety and efficacy of an existing therapy. Rolling Review provides a drug company the opportunity to submit completed sections of its NDA for review by the FDA. Typically, NDA reviews do not commence until the drug company has submitted the entire application to the FDA. Through the FTD, the FDA attempts to ensure that questions raised during the drug development process are resolved quickly, often leading to earlier approval and increased access for patients.
Current Treatments for Gliomas and Glioblastoma Multiforme
Gliomas are a type of Central Nervous System (“CNS”) tumor that arises from glial cells in the brain or spine. Glial cells are the cells surrounding nerves. Their primary function is to provide support and protection for neurons in the CNS.
Common symptoms of GBM include headaches, seizures, nausea, weakness, paralysis and personality or cognitive changes such as loss of speech or difficulty in thinking clearly. GBM progresses quickly and patients’ conditions deteriorate rapidly progressing to death. The outlook for GBM patients is generally poor. The overall median survival in newly diagnosed GBM patients
7
with best available treatments is less than 15 months, and two-year and five-year survival rates are approximately 30% and 10%, respectively. Median overall survival in newly-diagnosed, unmethylated GBM patients is 12.2 months.
The recommended treatment regimen for GBM includes surgical resection to remove as much of the tumor as possible (“debulking”) followed by radiotherapy with concomitant and adjuvant chemotherapy with TMZ with or without tumor treating fields (“TTF”). GBM patients whose tumors exhibit an unmethylated promoter for the gene encoding the DNA repair enzyme MGMT, a biomarker correlated with resistance to TMZ, may be treated with radiation alone following surgery.
Patients with an unmethylated MGMT promoter have high levels of MGMT, a naturally-occurring DNA repair enzyme that repairs tumor-fighting lesions induced by TMZ thus allowing a patient’s tumor to continue to grow despite treatment, which leads to poor outcomes. Measurement of MGMT methylation status has become routine in clinical practice as biomarker that correlates with response to TMZ and patient outcomes in GBM.
Probability of GBM Patient Survival Correlated to Expression of MGMT Enzyme (Unmethylated promoter = High MGMT Expression and Significantly Shorter Survival)
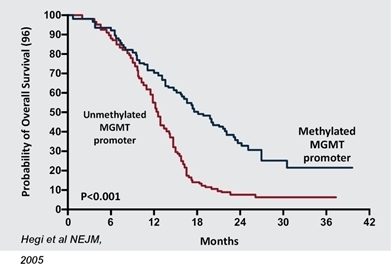
TTF (Optune®) is a non-invasive technique for adults with GBM. TTF uses alternating electrical fields to disrupt tumor cell division, or cause cell death, thereby preventing the tumor from growing or spreading as quickly. A clinical study reported that GBM patients treated with TTF combined with TMZ experienced longer survival than those treated with TMZ alone.
The majority of GBM patients’ tumors recur within 6 – 12 months of initial treatment. Experimental therapy through clinical studies is recommended under NCCN guidelines for eligible patients. NCCN guidelines also recommend treatment with systemic chemotherapy, such as lomustine (“CCNU”). For patients who are eligible for additional surgical debulking, local chemotherapy with carmustine (“BCNU”) wafers may be employed. CCNU and BCNU target the same DNA-site as TMZ and are also subject to MGMT-related resistance.
Avastin (Avastin®, an anti-VEGF antibody) recently received full approval in the US, Canada, Australia, and Japan as a single agent for patients with recurrent GBM following prior therapy. Avastin carries an FDA “black-box warning” related to severe, sometimes fatal, side effects such as gastrointestinal perforations, wound healing complications and hemorrhage. There are no data demonstrating an improvement in disease-related symptoms or increased survival for GBM patients treated with Avastin.
Recurrent GBM patients, especially those whose tumors progress following treatment with Avastin, have limited or no treatment options and a very poor prognosis. According to published literature, the median survival for GBM patients whose tumors progress following Avastin is less than five months.
8
VAL-083 Mechanism of Action
Chemotherapy forms the basis of treatment in nearly all cancers. We believe that VAL-083 may be effective in treating tumors exhibiting biological features that cause resistance to currently available chemotherapy, particularly for patients who have failed, or become resistant to, other treatment regimens.
Based on published research and our own data, the cytotoxic functional groups, and the mechanism of action of VAL-083 are functionally different from alkylating agents commonly used in the treatment of cancer. VAL-083 has previously demonstrated activity in cell-lines that are resistant to other types of chemotherapy. No evidence of cross-resistance has been reported in published clinical studies.
Our research suggests that VAL-083 attacks cancer cells via a unique mechanism of action that is distinct from other chemotherapies used in the treatment of cancer. Our data indicate that VAL-083 forms inter-strand crosslinks at the N7 position of guanine on the DNA of cancer cells. Our data also indicate that this crosslink forms rapidly and is not easily repaired by the cancer cell, resulting in cell-cycle arrest and lethal double-strand DNA breaks in cancer cells. VAL-083 readily crosses the blood brain barrier. Published preclinical and clinical research demonstrate that VAL-083 is absorbed more readily in tumor cells than in normal cells.
In vitro, our data also demonstrate that VAL-083’s distinct mechanism may be able to overcome drug resistance against a range of cancers. For example, VAL-083 is active against MGMT-unmethylated GBM cells which are resistant to treatment with TMZ and nitrosoureas. VAL-083 also retains a high level of activity in p53 mutated non-small cell lung cancer (“NSCLC”), ovarian cancer and medulloblastoma cell lines that are resistant to platinum-based chemotherapy.
Importantly, clinical activity against each of the tumors mentioned above was established in prior NCI-sponsored Phase 2 clinical studies. We believe that these historical clinical data and our own research support the development of VAL-083 as a potential new treatment for multiple types of cancer.
The main dose-limiting toxicity (“DLT”) related to the administration of VAL-083 in previous NCI-sponsored clinical studies and our own clinical studies is myelosuppression, particularly thrombocytopenia. Myelosuppression, including thrombocytopenia, is a common side effect of chemotherapy. Myelosuppression is the decrease in cells responsible for providing immunity, carrying oxygen, and causing normal blood clotting. Thrombocytopenia is a reduction in platelet counts which assist in blood clotting. Modern medicine allows for better management of myelosuppressive side effects. We believe this offers the potential opportunity to improve upon the drug’s already established efficacy profile by substantially increasing the dose of VAL-083 that can be safely administered to cancer patients.
There is no evidence of lung, liver, or kidney toxicity even with prolonged treatment by VAL-083. Data from the Chinese market where the drug has been approved for more than 15 years supports the safety findings of the NCI studies.
9
VAL-083 is Active Independent of MGMT
We have presented data at several peer reviewed meetings demonstrating that VAL-083 is active independent of MGMT resistance in GBM cell lines and other CNS tumor cells. Our research, along with that of others, demonstrates that VAL-083’s unique cytotoxic mechanism forms DNA cross-links at the N 7position of guanine and retains cytotoxic activity independent of MGMT expression in vitro. Our studies demonstrate that VAL-083 has more potent activity against brain tumor cells in comparison to TMZ and overcomes resistance associated with MGMT, suggesting the potential to surpass the current standard-of-care in the treatment of GBM.
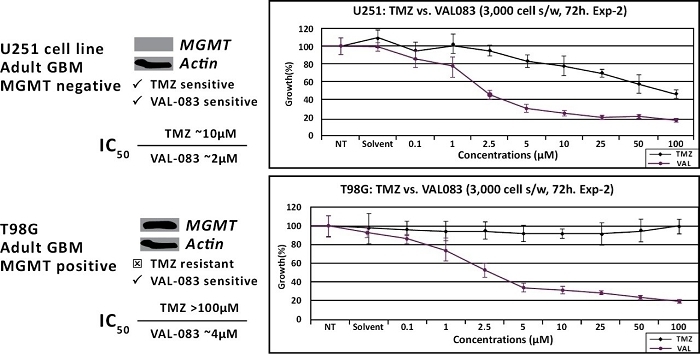
In addition, historical NCI clinical study data and our own research support the activity of VAL-083 as a potentiator of radiotherapy. Radiotherapy in combination with TMZ is the current standard of care in the treatment of newly diagnosed GBM. Our research demonstrates that TMZ and radiotherapy are ineffective against GBM cells exhibiting a high expression of MGMT, whereas VAL-083 potentiates the tumor-killing effect of radiation independent of MGMT expression. Furthermore, the combination of VAL-083 and radiation has been demonstrated to be active against GBM cancer stem cells (“CSCs”) in vitro. CSCs are often resistant to chemotherapy and form the basis for tumor recurrence and metastasis. GBM CSCs display strong resistance to TMZ, even where MGMT expression is low. However, our data demonstrates that GBM CSCs are susceptible to VAL-083 independent of MGMT expression.
Other Indications for VAL-083—Potential Future Opportunities
VAL-083 in Ovarian Cancer
With the FDA approval for our investigational new drug (“IND”) application for ovarian cancer, we have future plans for a phase 1/2, open-label, multicenter study of VAL-083 in patients with Recurrent Platinum Resistant Ovarian Cancer (“REPROVe”). Platinum-based chemotherapy is the standard-of-care in the treatment of ovarian cancer. Nearly all ovarian cancer patients eventually become resistant to platinum (“Pt”) based chemotherapy leading to treatment failure and poor patient outcomes. We have demonstrated that VAL-083 is active against Pt-resistant ovarian cancer in vitro. However, based on ongoing evaluation and input from our ovarian cancer advisory board, we are reassessing the development of VAL-083 for the treatment of ovarian cancer. We are in the process of evaluating the best path forward in ovarian cancer and are evaluating strategic options, including the potential combination of VAL-083 with PARP inhibitors. As a result, we have inactivated the IND while we explore alternative study designs.
VAL-083 in Lung Cancer
Lung cancer is a leading cause of cancer death around the world and effective treatment for lung cancer remains a significant global unmet need despite advances in therapy. Incidence of lung cancer in the United States is approximately 47 per 100,000 with the majority (85%) being NSCLC, the most common type of lung cancer. Globally, the market for lung cancer treatment may exceed $24 billion by 2033 according to a report published by Evaluate Pharma.
10
The activity of VAL-083 against solid tumors, including lung cancer, has been established in both preclinical and human clinical studies conducted by the NCI. We have developed nonclinical data to support the utility of VAL-083 in the modern treatment of lung cancer. In an established murine xenograft model of NSCLC, the activity of VAL-083 was compared to standard platinum-based therapy with cisplatin against human NSCLC cell lines A549 (TKI-sensitive) and H1975 (TKI-resistant). In the study, VAL-083 demonstrated superior efficacy and safety in the treatment of TKI-susceptible (A549) tumors and in TKI-resistant (H1975) tumors.
Central Nervous System Metastases of Solid Tumors
The successful management of systemic tumors by modern targeted therapies has led to increased incidence of mortality due to CNS metastases of lung cancer and other solid tumors. In June 2013, we split our Phase 1/2 clinical study protocol into two separate studies: one focusing solely on refractory GBM and the other focusing on secondary brain cancers caused by other tumors that have spread to the brain.
Based on historical clinical activity and our own research, we believe that VAL-083 may be suitable for the treatment of patients with CNS metastases who currently have limited treatment options. Subject to the availability of financial and operating resources, we plan to develop a separate protocol for the continued exploration of VAL-083 in patients with secondary brain cancer caused by a solid tumor spreading to the brain.
Pediatric Brain Tumors
Tumors of the brain and spine make up approximately 20 percent of all childhood cancers and they are the second most common form of childhood cancer after leukemia.
The activity of VAL-083 against childhood and adolescent brain tumors has been established in both preclinical and human clinical studies conducted by the NCI. We have presented data indicating that VAL-083 offers potential therapeutic alternatives for the treatment of pediatric brain tumors including SHH-p53 mutated medulloblastoma. In March 2016, the FDA granted orphan drug designation for the use of VAL-083 in the treatment of medulloblastoma. Subject to the availability of resources, we intend to collaborate with leading academic researchers for the continued exploration of VAL-083 as a potential treatment of childhood brain tumors.
Additional Indications for VAL-083
In historical studies sponsored by the NCI in the United States, VAL-083 exhibited clinical activity against a range of tumor types including central nervous system tumors, solid tumors, and hematologic malignancies. We have gathered nonclinical data supporting the activity of VAL-083 in different types of cancer that are resistant to modern targeted therapies and we believe that the unique cytotoxic mechanism of VAL-083 may provide benefit to patients in a range of indications. We intend to continue to research these opportunities, and if appropriate, expand our clinical development efforts to include additional indications.
VAL-083 Target Markets
VAL-083 target markets |
|
Estimated |
Glioblastoma multiforme by 2027 |
|
>$1.0B1 |
Ovarian cancer by 2028 |
|
>$6.0B1 |
Non-small cell lung cancer by 2027 |
|
>$22.0B2 |
Sources: 1 - GlobalData; 2 - iHealthcareAnalyst.
DNA-targeting agents such as alkylating agents or platinum-based chemotherapy form the mainstay of chemotherapy treatments used in the treatment of cancers. For example, TMZ had peak annual sales of $1.1 billion in 2010, while bendamustine, had peak annual sales of $0.8 billion in 2014.
We believe VAL-083 is a first-in-class DNA targeting agent with a novel mechanism of action. VAL-083’s anti-cancer activity was established in a range of tumor types in prior NCI-sponsored clinical studies. Based on this novel mechanism, we have demonstrated that the anti-cancer activity is maintained against tumor cells that are resistant to other DNA-targeting agents. We believe this positions VAL-083 as a potential chemotherapy-of-choice for patients whose tumors are resistant to current standard-of-care chemotherapy in orphan and major cancer indications.
11
Our ongoing research and development activities are focused on indications where VAL-083 demonstrated promising activity in prior NCI-sponsored studies and where our research suggests an opportunity to address significant unmet medical needs due to the failure of existing treatments.
Glioblastoma Multiforme
Newly diagnosed patients suffering from GBM are initially treated through invasive brain surgery, although disease progression following surgical resection is nearly 100%. Temodar® in combination with radiation is the front-line therapy for GBM following surgery. Global revenues of branded Temodar reached $1.1 billion in 2010. Approximately 60% of GBM patients treated with Temodar® experience tumor progression within one year. Median overall survival in newly-diagnosed, unmethylated GBM patients is 12.2 months.
Bevacizumab (Avastin®) has been approved for the treatment of GBM in patients failing Temodar®. In clinical studies, approximately 20% of patients failing Temodar® respond to Avastin® therapy and no improvement in median survival was reported.
The market for refractory (Avastin-failed) GBM is limited to those jurisdictions where Avastin is approved for the treatment of GBM. The United States, Canada, Australia, Japan and Switzerland represent the major markets where Avastin is used in the treatment of GBM.
Based on a November 2018 report from GlobalData, we believe there is a projected market opportunity for GBM of approximately $800 million, estimated to reach approximately $1.8 billion by 2027.
REM-001
Background
Through REM-001, we are developing our PDT for the treatment of rare, unmet medical needs. REM-001 (rostaporfin injectable emulsion) is a second-generation photosensitizer formulated as an intravenous, isotonic, isosmotic lipid emulsion containing the photo-activated active ingredient tin ethyl etiopurpurin ("SnET2"), also known as rostaporfin. PDT is a two-stage light-based treatment modality that utilizes a photosensitizing drug and light energy. The tumor-destroying effect of PDT is based on photo-oxidation. The process involves the administration of a photosensitizing drug that preferentially localizes to, and is retained by, tumor cells and tumor neovasculature. Administration of local light illumination to the tumor activates the photosensitizing drug which is inactive unless exposed to a specific range of light. Ideally, the light is delivered at the specific wavelength that allows for maximum absorption by the photosensitizing drug, as well as optimal tissue penetration. The light activation initiates a cascade of energy transfer events in which the excited photosensitizer reacts with cellular oxygen to generate reactive oxygen species.
REM-001 therapy is a combination therapy, comprising three components: a laser light source, a light delivery device, and the drug REM-001 (collectively the "REM-001 Therapy"). In use, REM-001 is first administered by intravenous infusion and allowed to distribute within the body and be taken up by the tumors. Tumors are then illuminated with light using the light delivery device which is attached to the laser light source (the KINTARA 665 Laser System), so that the accumulated REM-001 can be activated for the desired clinical effect.
Our lead indication for REM-001 is CMBC which is a disease that may strike individuals with advanced breast cancer and for which effective treatment options are limited. In four Phase 2 and/or Phase 3 clinical studies in CMBC patients, primarily targeting patients who had previously received chemotherapy and failed radiation therapy, REM-001 Therapy was able to reduce, or eliminate, a substantial number of the treated CMBC tumors. Specifically, our analysis of the data collected from these studies indicates that in approximately 80% of evaluable tumor sites treated with REM-001 Therapy, there was a complete response; meaning that follow-up clinical assessments indicated no visible evidence of the tumor remaining. We believe clinical data indicates that REM-001 Therapy holds promise as a treatment to locally eliminate, or slow the growth of, treated cutaneous cancerous tumors in this difficult-to-treat patient population.
Based on previous data, we believe REM-001 Therapy provides promising safety and efficacy in CMBC patients and that, taken together, these results provide strong support for REM-001 Therapy as a potential therapy for this disease. Furthermore, we believe the approvable letter previously granted to Miravant Medical Technologies Inc. ("Miravant") with respect to its NDA for REM-001 in an aspect of Age-related Macular Degeneration ("AMD") may indicate that many of the elements required for approval have already been completed for REM-001.
12
Numerous approaches have been utilized to treat CMBC patients, including various forms of chemotherapy, radiation therapy, surgical excision, hyperthermia, cryotherapy, electro-chemotherapy, topical drugs, and intra-lesional chemotherapy injections. However, for the most part, we believe that these therapies are often inadequate given the limited efficacy, toxicities and/or side effects of each. We believe our REM-001 Therapy has several advantages for this indication: it can be highly directed to the tumor site, has minimal systemic effects or normal tissue toxicities, can be used in conjunction with other therapies, and can be periodically repeated.
We believe REM-001 Therapy also may hold promise as a treatment for cutaneous metastatic cancers other than CMBC, as well as locally-advanced basal cell cancer such as often occurs in patients with BCCNS and cutaneously recurrent basal cell cancer. On January 16, 2018, the FDA granted our request that SnET2 (the active pharmaceutical ingredient in REM-001) be designated as an orphan drug for treatment of BCCNS.
In addition, we believe REM-001 Therapy also holds promise for certain cardiovascular conditions, including de novo treatment of cardiovascular access sites in hemodialysis patients to ameliorate current high failure rates. We hold an orphan drug designation for SnET2 for the prevention of access graft failure in hemodialysis patients. We have been working to further develop this indication, including engaging with a key opinion leader in this area and submitting a National Institute of Health grant proposal for late-stage preclinical research that we believe could lead directly to an IND and clinical study. On July 17, 2020, we received notification that that grant had been awarded.
REM-001 Regulatory Filings
The IND for REM-001 Therapy, IND 39,940, was filed in June 1992 with the FDA’s Division of Oncology and Pulmonary Drug Products. On August 9, 2022, we announced that we had received a Study May Proceed letter from the FDA to begin our 15-patient study evaluating REM-001 PDT for the treatment of CMBC.
Clinical Development Plans
CMBC
Our plan is to conduct an initial open-label, 15-patient study in CMBC to confirm planned dose and optimized study design followed by a Phase 3 clinical study in CMBC. We have received a Study May Proceed letter from the FDA to begin our 15-patient study evaluating REM-001 PDT for the treatment of CMBC. At this time, we estimate the necessary pivotal study design will be a Phase 3 multi-center study that would enroll CMBC patients who have received prior radiation therapy and chemotherapy.
Our plan is to use new lasers that are functionally equivalent to the lasers used in previous studies. Our laser is a portable solid-state diode laser system that is intended for use in PDT as the source of photoactivation of Rostaporfin for the treatment of subjects with cutaneous cancer lesions. Our laser system consists of the Kintara 665 laser with a fiber-coupled illuminator. In the case of cutaneous treatment, such as with CMBC, the light delivery device consists of an optical fiber which has a modified end to allow it to deliver a uniform light treatment field to the tumor. We have had clinical light delivery devices built by a contract medical device manufacturer using the previous basic design and tested to the same performance specifications as used previously.
The REM-001 Drug
REM-001 is a light activated photosensitizer drug used in PDT. During light activation, photosensitizer drugs act as a catalyst and absorb light energy which they transfer to surrounding oxygen-containing molecules to create reactive oxygen species (“ROS”). ROS can initiate various biological mechanisms of action:
13
REM-001 is a second-generation photosensitizer drug designed with the following attributes to overcome several of the shortcomings of earlier, first generation photosensitizer drugs:
REM-001 Safety and Toxicology
PDT carries what we believe is an inherent safety advantage since it uses photosensitizer compounds that are largely inactive except when they are being illuminated by intense light at specific wavelengths. Nevertheless, drug molecules, including photosensitizer molecules, can carry safety or toxicology risks on their own. REM-001 has previously undergone preclinical and clinical studies throughout its development cycle and has undergone certain tests typically required for FDA drug approval. REM-001 has been safely administered to over 1,100 patients in prior clinical studies. Most significantly, REM-001 has been previously reviewed by the FDA as part of the NDA submitted by Miravant for the use of REM-001 to treat an aspect of AMD, a non-CMBC indication. Following that review, the FDA granted an approvable letter for REM-001 in an aspect of AMD in 2004, with final approval contingent on, among other things, the successful completion of a Phase 3 study. While not definitive, we believe this letter, along with feedback we received from FDA meetings, indicates that it is unlikely that there will be significant safety or toxicology issues associated with REM-001 that would ultimately prevent marketing approval.
Based on our review of previous clinical data of CMBC studies, pain was the most common treatment-related adverse event experienced by patients in these studies. The second most common safety issue experienced with REM-001 was a transient photosensitivity, meaning extended exposure in bright light and direct sunlight should be avoided. Transient photosensitivity occurs with all photosensitizers to some degree. We believe this issue can be addressed by minimizing one’s exposure to bright light and sunlight for two to four weeks after treatment. In general, the potentially treatment-related adverse events observed in these CMBC studies were expected in nature (pain, edema, skin photosensitivity) and severity, and mostly resolved during the course of the studies.
REM-001 Therapy Target Markets
Our development plan for REM-001 Therapy is focused on the treatment of rare unmet needs in cancer, particularly those where the tumor can be accessed with a light delivery fiber device.
CMBC
While most internal cancers can metastasize to the skin, the internal cancer where this most commonly occurs is breast cancer. Radiotherapy is often used as an adjunctive therapy in breast cancer, in part to help prevent the development of local recurrences including CMBC. However, breast cancer survivors may still develop CMBC lesions, even over a decade after their original cancer treatment. In fact, physicians often watch for cutaneous (skin surface) metastases as a sign of breast cancer recurrence. A 2003 meta-analysis of approximately 20,000 cancer patients found that 24% of metastatic breast cancer patients included in the analysis had developed cutaneous metastases, which was the highest rate of skin metastases of any cancer type. Given that approximately 168,000 women in the U.S. suffer from metastatic breast cancer, we believe the prevalence of CMBC may approach 40,000 in the United States. In many cases of CMBC, surgical excision is not possible, so various standard cancer therapies, particularly radiotherapy or chemotherapy, are the first course of treatment. We believe these therapies are inadequate given the well-known dose limiting toxicities, limited efficacy, and/or side effects of each. We are not aware of any prospective clinical studies that have led to FDA approval of a therapy specifically for the treatment of CMBC and we do not expect any to be approved in the near future.
According to a market assessment from Charles River Associates (2018), there is an estimated market opportunity of approximately $500 million for the treatment of CMBC.
Cutaneous Metastatic Cancers
A meta-analysis has shown that approximately five percent of people with internal (non-melanoma, non-lymphatic, non-leukemic) cancers develop cutaneous metastatic tumors in their skin. Based on an estimated incidence of 1,500,000 such internal cancers in the United States, this means that the incidence of such cutaneous metastases is approximately 75,000 with a substantially higher prevalence given the fact that individuals often live with metastatic cancer for years. Regardless of the primary source of the cancer, these cutaneous metastatic tumors often begin as small skin nodules but, as the cancer spreads, more nodules form and can
14
eventually cover large areas of skin. With progression, the tumor field generally becomes more painful as tumors may grow larger and more numerous, ulcerate, bleed and carry a strong odor. Part of our goal is to treat these cutaneous tumors as early as possible to either cause them to be locally eliminated or to slow their growth sufficiently to reduce their late-stage development.
Basal Cell Carcinoma Nevus Syndrome
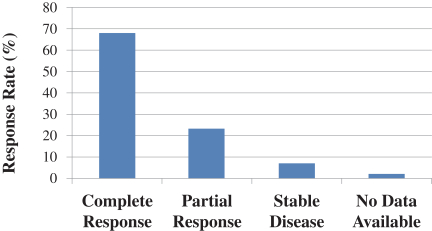
In addition to the clinical studies that Miravant conducted with REM-001 Therapy in CMBC, it also generated clinical data for patients with BCCNS who developed extensive basal cell carcinoma. BCCNS is a rare but serious condition that is often characterized by the formation of multiple and recurring cutaneous basal cell carcinoma lesions. According to Cancer.net, as of April 2020, approximately 1 in 40,000 individuals in the U.S. have an underlying genetic condition that causes BCCNS and approximately 90% of these have BCCNS and it has been recognized as an orphan indication by FDA. In a previous Phase 1/2 clinical study (CA001B), 14 patients with BCCNS were enrolled and treated with REM-001 Therapy using the same dosing conditions as were used in the CMBC studies. A total of 157 lesions were treated in these patients and showed a 91% overall response rate. This was composed of a 68% complete response rate (no remaining visible evidence of a lesion) and a 23% partial response rate (lesion was reduced in size by more than 50%). In addition, 7% of lesions had stable disease (any increase in lesion size was less than 25%). The various response rates are shown in the graph below and are similar to the results seen in CMBC patients as we would expect. Based on these results we requested, and were granted, an orphan drug designation for SnET2.
Until the FDA approval of the drugs Odomzo (2015) and Erivedge (2012) treatment options for these BCCNS patients were very limited. However, we believe that, based on their package inserts, Odomzo and Erivedge have dose limiting toxicity profiles which are broader in scope than the primarily transient adverse effects observed to-date with REM-001 Therapy. We believe that the potential toxicity limitations related to the existing therapies for BCCNS, plus the positive initial Phase 1/2 data generated in clinical studies with REM-001 Therapy, suggest that REM-001 Therapy could be a viable alternative in treating recurrent basal cell carcinoma in BCCNS patients.
Current and Experimental Treatments for CMBC
As with many cancers, the current standard treatment for CMBC is surgical excision. However, this is often not feasible due to the extent of the tumor field or the condition of the skin, particularly in patients who have had radiation therapy. A number of other therapies have been used on patients with CMBC, including various forms of chemotherapy, radiation therapy, hyperthermia, cryotherapy, electro-chemotherapy, topical drugs and intra-lesional chemotherapy injections. Researchers have also attempted to combine therapies in an effort to improve efficacy. However, we believe that these therapies are often inadequate given the limited efficacy, toxicities and/or side effects of each. The side effects associated with therapies may be particularly difficult for patients who may have already experienced extensive surgery along with a full course of radiation and/or systemic chemotherapy. Also, the fact that CMBC tumors continue to develop following these therapies is a signal that the tumor cells may have developed a resistance to some of these approaches. Based on our discussions with clinicians and literature reviews, and the March 3, 2017, response from FDA, we believe that treatment of unresectable CMBC tumors is a largely unmet medical need, particularly in patients who have already received extensive radiation and chemotherapy.
15
Clinical Results in CMBC
While we have not conducted any clinical studies, we have undertaken an analysis of the Phase 1 and four Phase 2 and/or Phase 3 CMBC clinical studies done previously with REM-001 Therapy by Miravant. We have concluded that in these studies REM-001 Therapy provided higher tumor response rates than are generally seen with alternative CMBC treatments. However, this program was discontinued in 1998. Our review of clinical records further indicates that following this decision, Miravant continued to monitor patients in the CMBC studies and collected data as required by protocol, but they conducted no further treatment of CMBC patients with REM-001 Therapy. We believe that Miravant primarily chose to discontinue this program in order to focus its REM-001 development efforts on an aspect of “wet” AMD.
Phase 2/3 Studies
After completion of the Phase 1 dose finding study, four Phase 2/3 studies were conducted with REM-001 Therapy for the treatment of CMBC as summarized below. These studies all used the same dosimetry as described above and most of the patients had been previously treated with radiation therapy and chemotherapy. The light delivery devices used in these studies were the ML1-0400 or the functionally equivalent ML2-0400. The laser light source used in three of the studies was the Miravant DD2 laser and one study used the KTP model laser manufactured by LaserScope. Each study was conducted under the cancer IND using Good Clinical Practices with safety and efficacy data collected accordingly. In connection with our acquisition of the Miravant assets, ownership of that IND has been transferred to us.
The table below summarizes the CMBC Studies. Studies CA008, CA009 and CA019 required that the patients enrolled had received prior radiation therapy. Study CA013 did not have this specific inclusion requirement but our review of the data indicates that at least 50 of the 56 patients in CA013 had received prior radiation therapy. A second difference across the studies is that studies CA008, CA009 and CA019 had a 24-week follow-up period while study CA013 had a 52-week follow-up period. Also, in studies CA008 and CA009 two tumor lesions on each patient were randomly selected as controls and did not receive light activation. CA013 was conducted in Europe by a corporate partner of Miravant. Beyond these differences and those device differences noted above, we believe there were no other substantive differences between the studies and that all studies enrolled similar patients.
16
Table of Phase 2 and/or 3 CMBC Studies
(Note: SnET2 is now called REM-001)
Trial Title |
|
Phase |
|
|
Location |
|
Total |
|
|
Total |
|
|
Included |
|||
CA008: Open-Label Randomized No Treatment |
|
2/3 |
|
|
U.S. |
|
|
32 |
|
|
|
32 |
|
|
Yes |
|
CA009: Open-Label Randomized No Treatment |
|
2/3 |
|
|
U.S. |
|
|
36 |
|
|
|
36 |
|
|
Yes |
|
CA013: Multinational, Open-Label Study of Single Dose |
|
|
2 |
|
|
Europe |
|
|
56 |
|
|
|
50 |
|
|
No |
CA019: Open-Label Study of Single Dose Tin Ethyl |
|
|
3 |
|
|
U.S. |
|
|
25 |
|
|
|
25 |
|
|
No |
The primary endpoints for studies CA008 and CA009 were objective tumor response rate, quality-of-life change, device performance and patient safety. Our review of the tumor response rate and quality-of-life endpoints indicated they were defined as follows:
No significant device failures were observed in either study. Secondary endpoints in CA008 and CA009 were patient disease burden, duration of response and patient pain assessment. Previous analysis indicated, for patients for which data was available, there was a treatment benefit in disease burden (p = 0.0017 for CA008, p = 0.0020 for CA009) and duration of response (p < 0.001 for CA008, not significant in CA009) when comparing treated and control lesions. In terms of pain, there was no significant change in pain in CA008 and a treatment related increase in pain at 4 Weeks post-treatment in CA009. Treatment related pain, particularly during the first month after treatment, was the most commonly reported adverse event and was often treated with analgesics.
17
Studies CA013 and CA019 used similar endpoints with one notable exception. Tumor Response as Measured by Paired Response was not possible in these studies since this measurement relies on control lesions and CA013 and CA019 did not include controls. Miravant did not conduct an efficacy analysis of these two studies but we have conducted an analysis of the Quality of Life and Clinical Success endpoints used in the pivotal CA008 and CA009 studies. Results from that analysis are shown in the following table:
|
|
Clinical Success |
|
24 Week Quality of Life Change |
|
|||||||||||||||
Study |
|
Eligible |
|
|
Average |
|
|
95% |
|
Eligible |
|
|
Mean ± SD |
|
P value |
|
||||
CA013 |
|
|
32 |
|
|
|
88 |
% |
|
71% - 97% |
|
|
16 |
|
|
1.3 ± 3.6 |
|
|
1.00 |
|
CA019 |
|
|
18 |
|
|
|
83 |
% |
|
45% - 86% |
|
|
11 |
|
|
2.5 ± 4.7 |
|
|
1.00 |
|
The most common adverse events seen in these four studies (CA008, CA009, CA013, CA019) were pain and photosensitivity, both of which are expected with this therapy. In the four studies there were a total of 17 serious adverse events (SAE’s) that were judged by investigators to be possibly, probably or definitely related to treatment. None of these were classified by the investigator as life threatening and none resulted in death. Of these 17 SAE’s, eight were related to necrosis of the treated lesions, three were related to treatment field infection, four were treatment related pain, one was a photosensitivity skin reaction and one was an allergic reaction.
We believe that the data from these studies show that REM-001 Treatment is a promising therapy for CMBC. However, because there are no approved therapies for CMBC, we have no basis for comparing these results to existing therapies. Based on the FDA’s March 3, 2017 response, we believe the FDA will view these results as supportive data and our plan is to conduct a new pivotal Phase 3 study to support a new drug application.
The figure below shows the results of this initial preliminary analysis of the clinical data and depicts the percentage of evaluable lesions in each CMBC Study for which there was a complete response; i.e. where all visible clinical evidence of the tumor is gone after treatment with REM-001 Therapy.
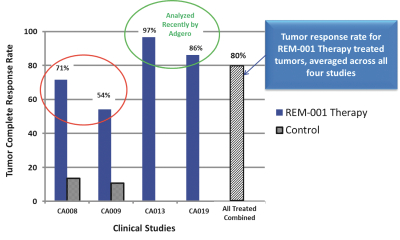
Manufacturing
VAL-083
VAL-083 is a small-molecule chemotherapeutic. Chemical synthesis of the active pharmaceutical ingredient (“API”) was initially established by the NCI. We have made improvements to this process and have obtained patents on these improvements. The current manufacturing process involves fewer than five synthetic steps.
VAL-083 drug product is a lyophilized (freeze-dried) formulation that is reconstituted for intravenous injection. We anticipate that overall cost of goods for an eventual commercial product will be similar to other injectable, small-molecule pharmaceuticals.
18
For our now-completed clinical study that was conducted in China, the supply of VAL-083 was provided through our collaboration with Guangxi Wuzhou Pharmaceutical Company. To-date, they have not achieved the quality of GMP manufacturing systems necessary to meet FDA manufacturing standards.
To address the need to meet FDA standards, we have engaged third-party contract manufacturers with the capabilities to establish the processes, procedures and quality systems necessary to meet U.S., Canadian, E.U. and other international manufacturing requirements in accordance with Good Manufacturing Practice (“cGMP”) regulations. We have used drug supply from these third-parties for our ongoing GCAR study.
We have developed and patented certain intellectual property related to quality controls that are used in the release of VAL-083 for our clinical studies.
REM-001
The manufacturing process for the API in REM-001 was developed over a ten-year period and we believe is now well established and suitable for commercial scale production. This process was also included as part of Miravant’s prior NDA for the use of REM-001 to treat an aspect of AMD, which underwent an FDA review where an approvable letter was granted. The final REM-001 drug product is a lipid-based formulation and was previously produced at a commercial scale by a contract manufacturer for use in Miravant’s previous clinical studies and commercialization activities. We do not own or operate manufacturing facilities for the production of REM-001, nor the laser light source, or light delivery device for use with REM-001 Therapy. We are dependent on third-party suppliers and manufacturing organizations for both commercial and clinical study supplies of all of our raw materials, the REM-001 drug substance, drug product and the REM-001 Therapy, laser light source, and light delivery device.
We have engaged a contract manufacturer who has manufactured the starting material for our API, and then manufactured two API lots under GMP and has stability testing ongoing. We have also engaged a contract manufacturer who has manufactured a drug product lot under GMP for use in our planned 15-patient clinical study. With the feedback from the FDA that we could utilize the existing supply of laser systems or devices that were functionally equivalent, an in-depth assessment was made to determine which pathway would be appropriate. It has been determined that the existing lasers that were utilized in the previous clinical studies will not be used in the current clinical studies. We engaged a third-party contract medical device manufacturer who has built new lasers and light-delivery devices. We have also engaged an affiliate of this manufacturer to train the clinical staff in the use of the units, provide regulatory support for the devices, and maintain the devices while being used in the study. We believe there are readily available supplies of all raw materials needed for the manufacture of REM-001 and the related required light device components to satisfy future requirements.
Research and Development Collaborations
Guangxi Wuzhou Pharmaceutical Company
Pursuant to a memorandum of understanding and collaboration agreement, dated October 25, 2012, we have established a strategic collaboration with Guangxi Wuzhou Pharmaceutical Company, a subsidiary of publicly traded Guangxi Wuzhou Zhongheng Group Co., Ltd. (SHG: 600252) (the “Guangxi Agreement”). VAL-083 is approved for the treatment of chronic myelogenous leukemia (“CML”) and lung cancer in China and Guangxi Wuzhou Pharmaceutical Company is the only manufacturer licensed by the CFDA to produce the product for the China market. Through the Guangxi Agreement, we have been provided with drug product for our now-completed Phase 2 study in China as well as for certain completed clinical studies in the United States. In addition, we have secured certain commercial rights in China.
Pursuant to the Guangxi Agreement, we have granted to Guangxi Wuzhou Pharmaceutical Company a royalty-free license to certain of our intellectual property for use in China as it relates to quality control and drug production methods for VAL-083. In addition, subject to successful agreement on definitive commercial terms, we have agreed that Guangxi Wuzhou Pharmaceutical Company will be our exclusive supplier of VAL-083 for clinical studies and commercial sales, subject to Guangxi Wuzhou Pharmaceutical Company obtaining and maintaining cGMP certification by the FDA, EMA or other applicable regulatory agencies, and Guangxi Wuzhou Pharmaceutical Company being able to meet volumes ordered by us. To-date, Guangxi Wuzhou Pharmaceutical Company has not achieved cGMP certification with respect to the manufacturing of VAL-083.
This Guangxi Agreement also provides us with certain exclusive commercial rights related to drug supply. Specifically, the Guangxi Agreement establishes an exclusive supply relationship between us and Guangxi Wuzhou Pharmaceutical Company for the Chinese market and all markets outside China. Guangxi Wuzhou Pharmaceutical Company agreed that it may not sell VAL-083 for markets outside of China to any other purchaser other than us, provided that, during the first three years following regulatory clearance for marketing of VAL-083 in a particular country or region, we meet proposed sales volumes set by Guangxi Wuzhou Pharmaceutical Company for the country or region. In addition, Guangxi Wuzhou Pharmaceutical Company granted us a pre-emptive right in China
19
(subject to our acceptance of proposed sales volume and prices) to purchase VAL-083 produced by Guangxi Wuzhou Pharmaceutical Company.
With respect to the Phase 3, or registration study, for GBM to be undertaken in China in order to ultimately commercialize VAL-083 in China, we are not under an obligation to participate in such a study. However, our participation in such a study in China, for the Chinese market, would be part of a larger negotiation process between us and Guangxi Wuzhou Pharmaceutical Company to determine how such a study would be conducted. We plan to execute a Phase 3 study and to seek approval for VAL-083 outside of China and we have no dependency, or obligations, to Guangxi Wuzhou Pharmaceutical Company with respect to studies we plan to conduct outside of China.
The term of the Guangxi Agreement (except as it relates to the exclusive rights in the China market) is indefinite, subject to termination upon written agreement of all parties, or if either party breaches any material term and fails to remedy such breach within 30 days of receipt of notice of the breach, or if any action to be taken thereunder is not agreed to by both parties, provided that such matter is referred to the chief executive officer of both parties, and they are unable to resolve such matter within 90 days. No payments have been made to date under the Guangxi Agreement to, or from, either Kintara or Guangxi Wuzhou Pharmaceutical Company.
St. Cloud Asset Purchase Agreement
Adgero acquired certain Miravant assets, including the REM-001 Therapy and the associated technology and intellectual property, through an asset purchase agreement with St. Cloud Investments, LLC (“St. Cloud”), dated November 26, 2012, as amended (the “St. Cloud Agreement”). In conjunction with the merger with Adgero which closed on August 19, 2020, we assumed the St. Cloud Agreement. St. Cloud was previously a Miravant creditor and acquired these Miravant assets pursuant to a foreclosure process St. Cloud completed under California law. Pursuant to the terms of the St. Cloud Agreement, we are obligated to make certain payments under the agreement.
As of June 30, 2022, the amounts still to be paid or owed under that agreement are as follows:
With respect to the $300,000 and $700,000 potential milestone payments referenced above (each a “Milestone Payment”), if either such Milestone Payment becomes payable, and in the event we elect to pay either such Milestone Payment in shares of our common stock, the value of the common stock will equal the price per share of the most recent financing, or, if we are considered to be a publicly-traded company, the average of the closing price per share of our common stock over the twenty (20) trading days following the first public announcement of the applicable event described above.
In addition, we must pay to St. Cloud and Steven Rychnovsky, PhD, in the aggregate, a royalty fee of six percent (6%) of net sales during the royalty term on a country-by-country and product-by-product basis with St. Cloud receiving a royalty rate of four and eight tenths percent (4.8%) and Steven Rychnovsky, PhD, receiving a royalty of one and two tenths percent (1.2%). The royalty term for a product commences on the first commercial sale of the product, such as REM-001 Therapy, in any country, and the royalty fee must be paid within 30 days of each calendar quarter during which revenue is collected. The royalty term terminates on the later of (i) the invalidation, revocation, lapse or expiration of the last to expire valid claim on any patent acquired in the St. Cloud Agreement that would be infringed by the sale of the product in the country where the commercial sale takes place or (ii) the expiration of the period for which we hold exclusive marketing rights of the product in the country, if we were granted those rights under the St. Cloud Agreement.
Patents and Proprietary Rights
Our success will depend in part on our ability to protect our existing product candidates and the products we acquire or license by obtaining and maintaining a strong proprietary position. To develop and maintain our position, we intend to continue relying upon the validity and enforceability of our patents patent protection, orphan drug status, Hatch-Waxman exclusivity, trade secrets, know-how, continuing technological innovations and licensing opportunities.
20
There is no guarantee that patents will be granted with respect to any patent applications we may submit, own or license in the future, nor can we be sure that any of our existing patents or any patents we may own or license in the future will be useful in protecting our technology.
VAL-083
We have filed patent applications claiming the use of, and improvements related to VAL-083. Our patent filings also include proposed treatment regimens, improvements to the manufacturing process, formulation and composition of the active pharmaceutical ingredient, and finished dosage forms of VAL-083. We are prosecuting and maintaining our patent applications in the United States and other jurisdictions which we deem important for the potential commercial success of VAL-083.
Our patents and patent applications for VAL-083 can be summarized in fourteen series as follows:
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent No. 8,563,758 |
|
Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol |
|
|
United States Patent No. 8,921,585 |
|
Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol |
|
|
United States Patent No. 9,085,544 |
|
Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol |
|
|
United States Patent No. 9,630,938 |
|
Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol |
|
|
PCT Patent Application Serial No. PCT/US2011/048032 |
|
Method Of Synthesis Of Substituted Hexitols Such As Dianhydrogalactitol. Patents granted in various countries. |
|
2031 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent No. 9,066,918 |
|
Compositions And Methods To Improve The Therapeutic Benefit Of Suboptimally Administered Chemical Compounds Including Substituted Hexitols Such As Dianhydrogalactitol And Diacetyldianhydrogalactitol |
|
|
United States Patent No. 9,901,563 |
|
Compositions And Methods To Improve The Therapeutic Benefit Of Suboptimally Administered Chemical Compounds Including Substituted Hexitols Such As Dianhydrogalactitol And Diacetyldianhydrogalactitol |
|
|
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent No. 9,759,698 |
|
Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol |
|
|
United States Patent No. 10,145,824 |
|
Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol |
|
|
United States Patent No. 9,029,164 |
|
Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol |
|
|
PCT Patent Application Serial No. PCT/IB2013/000793 |
|
Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol. Patents granted in various countries. |
|
2033 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
PCT Patent Application Serial No. PCT/US2014/066087 |
|
Improved Analytical Methods For Analyzing And Determining Impurities In Dianhydrogalactitol |
|
2034 |
21
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent No. 11,234,955 |
|
Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma |
|
|
United States Patent No. 9,687,466 |
|
Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma |
|
|
United States Patent No. 10,201,521 |
|
Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma |
|
|
United States Patent Application Serial No. 17/583,141 |
|
Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma |
|
|
PCT Patent Application Serial No. PCT/US2013/022505 |
|
Use Of Substituted Hexitols Including Dianhydrogalactitol And Analogs To Treat Neoplastic Disease And Cancer Stem Cells Including Glioblastoma Multiforme And Medulloblastoma. National phase applications pending and granted in various countries. |
|
2033 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent No. 9,814,693 |
|
Veterinary Use Of Dianhydrogalactitol, Diacetyldianhydrogalactitol, And Dibromodulcitol To Treat Malignancies |
|
|
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 17/177,665 |
|
Methods For Treating Tyrosine-Kinase-Inhibitor-Resistant Malignancies In Patients With Genetic Polymorphisms Or Ahi1 Dysregulations Or Mutations Employing Dianhydrogalactitol, Diacetyldianhydrogalactitol, Dibromodulcitol, Or Analogs Or Derivatives Thereof |
|
|
PCT Patent Application Serial No. PCT/US2013/047320 |
|
Methods For Treating Tyrosine-Kinase-Inhibitor-Resistant Malignancies In Patients With Genetic Polymorphisms Or Ahi1 Dysregulations Or Mutations Employing Dianhydrogalactitol, Diacetyldianhydrogalactitol, Dibromodulcitol, Or Analogs Or Derivatives Thereof. National phase applications pending and granted in various countries. |
|
2033 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent No. 11,026,914 |
|
Use Of Dianhydrogalactitol And Analogs And Derivatives Thereof To Treat Recurrent Malignant Glioma Or Progressive Secondary Brain Tumor |
|
|
PCT Application Serial No. PCT/US2014/040461 |
|
Use Of Dianhydrogalactitol And Analogs And Derivatives Thereof To Treat Recurrent Malignant Glioma Or Progressive Secondary Brain Tumor. National phase applications pending and granted in various countries. |
|
2034 |
22
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 14/710,240 |
|
Use of Dianhydrogalactitol and Analogs or Derivatives Thereof in Combination With Platinum-Containing Antineoplastic Agents to Treat Non Small-Cell Carcinoma of the Lung and Brain Metastases |
|
|
PCT Patent Application Serial No. PCT/US2015/024462 |
|
Use of Dianhydrogalactitol and Analogs or Derivatives Thereof to Treat Non-Small Cell Carcinoma of the Lung and Ovarian Cancer. National phase applications pending and granted in various countries. |
|
2035 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 15/525,933 |
|
Dianhydrogalactitol Together with Radiation to Treat Non-Small-Cell Carcinoma of the Lung and Glioblastoma Multiforme. |
|
|
PCT Patent Application Serial No. PCT/US2015/059814 |
|
Dianhydrogalactitol Together with Radiation to Treat Non-Small-Cell Carcinoma of the Lung and Glioblastoma Multiforme. National phase applications pending and granted in various countries. |
|
2035 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 15/759,104 |
|
Use of Dianhydrogalactitol And Derivatives Thereof in the Treatment of Glioblastoma, Lung Cancer and Ovarian Cancer. |
|
|
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 15/771,631 |
|
Use of Dianhydrogalactitol or Derivatives and Analogs Thereof for Treatment of Pediatric Central Nervous System Malignancies. |
|
|
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent No. 10,591,445 |
|
Methods for analysis and Resolution of Preparations of Dianhydrogalactitol and Derivatives and Analogs Thereof. |
|
|
PCT Patent Application Serial No. |
|
Methods for analysis and Resolution of Preparations of Dianhydrogalactitol and Derivatives and Analogs Thereof. National phase applications pending and granted in various countries. |
|
2036 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 16/609,721 |
|
Use of Dianhydrogalactitol and Analogs and Derivatives in Combination VEGF inhibitors to Treat Cancer |
|
|
|
|
|
|
|
23
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 16/489,122 |
|
Use of Dianhydrogalactitol and Analogs and Derivatives in Combination with a P53 Modulator or a PARP Inhibitor |
|
|
PCT Patent Application Serial No. |
|
Use of Dianhydrogalactitol and Analogs and Derivatives in Combination with a P53 Modulator or a PARP Inhibitor. National phase applications pending. |
|
2038 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 16/768,827 |
|
Use of Dianhydrogalactitol or Derivatives and Analogs Thereof to Treat Diffuse Intrinsic Pontine Glioma. |
|
|
In addition to patent protection, we may also seek orphan drug status whenever it is available. If a product which has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and Canada, and 10 years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for a different clinical indication.
VAL-083 has been granted protection under the Orphan Drug Act by the FDA and the European Medicines Agency ("EMA") for the treatment of gliomas, including GBM. The FDA has also granted Orphan Drug protection to VAL-083 for the treatment of medulloblastoma and ovarian cancer.
In February 2012, the FDA granted orphan drug status to VAL-083 for the treatment of glioma. In January 2013, the EMA also granted orphan drug protection to VAL-083 for the treatment of glioma. In the spring of 2016, the FDA Office of Orphan Products Development granted orphan drug designations to VAL-083 for the treatment of ovarian cancer and medulloblastoma.
In addition to our patents and orphan drug protection, we intend to rely on the Hatch-Waxman Amendments for five years of data exclusivity for VAL-083.Under the Hatch-Waxman Amendments, newly approved drugs and indications benefit from a statutory period of non-patent marketing exclusivity. These amendments provide five-year data exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active ingredient. The Hatch-Waxman Amendments prohibit the approval of an abbreviated new drug application, also known as an ANDA or generic drug application, during the five-year exclusive period if no patent is listed. If there is a patent listed and the ANDA applicant certifies that the NDA holder’s listed patent for the product is invalid or will not be infringed, the ANDA can be submitted four years after NDA approval. Protection under the Hatch-Waxman Amendments will not prevent the filing or approval of another full NDA; however, the applicant would be required to conduct its own pre-clinical studies and adequate and well-controlled clinical studies to demonstrate safety and effectiveness. The Hatch-Waxman Amendments also provide three years of data exclusivity for the approval of NDAs with new clinical studies for previously approved drugs and supplemental NDAs, for example, for new indications, dosages or strengths of an existing drug, if new clinical investigations were conducted by or on behalf of the sponsor and were essential to the approval of the application. This three-year exclusivity covers only the new changes associated with the supplemental NDA and does not prohibit the FDA from approving ANDAs for drugs containing the original active ingredient.
We also rely on trade secret protection for our confidential and proprietary information. We believe that the substantial costs and resources required to develop technological innovations, such as the manufacturing processes associated with VAL-083, will help us to protect the competitive advantage of our product candidate.
The protection of intellectual property rights in China (where VAL-083 is manufactured pursuant to a collaboration agreement with the only manufacturer presently licensed by the CFDA to produce the product for the China market, and where VAL-03 is approved for the treatment of CML and lung cancer) is relatively weak compared to the United States, which may negatively affect our ability to generate revenue from VAL-083 in China.
Our policy is to require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of employees and consultants, the agreements provide that all inventions conceived by the individual shall be our exclusive property.
24
REM-001
Our product pipeline for REM-001 is based on technology that was originally developed by Miravant. We acquired this technology, which includes scientific and regulatory data and product know-how, through an asset purchase agreement. We rely on trade secret protection for our confidential and proprietary information related to REM-001 and have filed patent applications to protect our intellectual property.
Our patent applications for REM-001 can be summarized as follows:
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 17/614,132 |
|
Methods for the production of Nickel (II) Etioporphyrin-I. |
|
|
PCT Patent Application Serial No. |
|
Methods for the production of Nickel (II) Etioporphyrin-I. |
|
2041 |
Patent or Patent Application No. |
|
Title |
|
Expiry |
United States Patent Application Serial No. 17/546,715 |
|
Methods for treating cutaneous metastatic cancers. |
|
|
PCT Patent Application Serial No. |
|
Methods for treating cutaneous metastatic cancers. |
|
2041 |
We own proprietary regulatory data for REM-001 which includes two INDs for use of REM-001 in oncology and ophthalmology, and one NDA for use of REM-001 to treat age-related macular degeneration ("AMD"). The FDA granted our request that tin ethyl etiopurpurin (the active pharmaceutical ingredient in REM-001) be designated as an orphan drug for treatment of basal cell carcinoma nevus syndrome ("BCCNS"). We also hold an orphan drug designation that was initially awarded to Miravant for tin ethyl etiopurpurin for the prevention of access graft disease in hemodialysis patients.
Government Regulation and Product Approval
Regulation by governmental authorities in the U.S. and other countries is a significant factor, affecting the cost and time of our research and product development activities, and will be a significant factor in the manufacture and marketing of any approved products. Our product candidates will require regulatory approval by governmental agencies prior to commercialization. In particular, our products are subject to rigorous preclinical and clinical testing and other approval requirements by the FDA and similar regulatory authorities in other countries. Various statutes and regulations also govern or influence the manufacturing, safety, reporting, labeling, transport and storage, record keeping and marketing of our products. The lengthy process of seeking these approvals, and the subsequent compliance with applicable statutes and regulations, require the expenditure of substantial resources. Any failure by us to obtain, or any delay in obtaining, the necessary regulatory approvals could harm our business.
The regulatory requirements relating to the testing, manufacturing and marketing of our products may change from time to time and this may impact our ability to conduct clinical studies and the ability of independent investigators to conduct their own research with support from us.
The clinical development, manufacturing and marketing of our products are subject to regulation by various authorities in the U.S., the E.U. and other countries, including, in the U.S., the FDA, in Canada, Health Canada, and, in the E.U., the EMA. The Federal Food, Drug, and Cosmetic Act, the Public Health Service Act in the U.S. and numerous directives, regulations, local laws and guidelines in Canada and the E.U. govern the testing, manufacture, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of our products. Product development and approval within these regulatory frameworks takes a number of years and involves the expenditure of substantial resources.
Regulatory approval will be required in all the major markets in which we seek to develop our products. At a minimum, approval requires the generation and evaluation of data relating to the quality, safety, and efficacy of an investigational product for its proposed use. The specific types of data required and the regulations relating to this data will differ depending on the territory, the drug involved, the proposed indication and the stage of development.
In general, new chemical entities are tested in animals until adequate evidence of safety is established to support the proposed clinical study protocol designs. Clinical studies for new products are typically conducted in three sequential phases that may overlap. In Phase 1, the initial introduction of the pharmaceutical into either healthy human volunteers or patients with the disease (20 to 50 subjects), the emphasis is on testing for safety (adverse effects), dosage tolerance, metabolism, distribution, excretion and clinical
25
pharmacology. Phase 2 involves studies in a limited patient population (50 to 200 patients) to determine the initial efficacy of the pharmaceutical for specific targeted indications, to determine dosage tolerance and optimal dosage and to identify possible adverse side effects and safety risks. Once a compound shows preliminary evidence of some effectiveness and is found to have an acceptable safety profile in Phase 2 evaluations, Phase 3 studies are undertaken to more fully evaluate clinical outcomes in a larger patient population in adequate and well-controlled studies designed to yield statistically sufficient clinical data to demonstrate efficacy and safety.
In the U.S., specific preclinical data, manufacturing and chemical data, as described above, need to be submitted to the FDA as part of an IND application, which, unless the FDA objects, will become effective 30 days following receipt by the FDA. Phase 1 studies in human volunteers may commence only after the application becomes effective. Prior regulatory approval for human healthy volunteer studies is also required in member states of the E.U. Currently, in each member state of the E.U., following successful completion of Phase 1 studies, data are submitted in summarized format to the applicable regulatory authority in the member state in respect of applications for the conduct of later Phase 2 studies. The regulatory authorities in the E.U. typically have between one and three months in which to raise any objections to the proposed study, and they often have the right to extend this review period at their discretion. In the U.S., following completion of Phase 1 studies, further submissions to regulatory authorities are necessary in relation to Phase 2 and 3 studies to update the existing IND.
Authorities may require additional data before allowing the studies to commence and could demand that the studies be discontinued at any time if there are significant safety issues. In addition to the regulatory review, studies involving human subjects must be approved by an independent body. The exact composition and responsibilities of this body will differ from country to country. In the U.S., for example, each study will be conducted under the auspices of an independent institutional review board ("IRB") at each institution at which the study is conducted. The IRB considers among other things, the design of the study, ethical factors, the privacy of protected health information as defined under the Health Insurance Portability and Accountability Act, the safety of the human subjects and the possible liability risk for the institution. Equivalent rules to protect subjects’ rights and welfare apply in each member state of the E.U. where one or more independent ethics committees, which typically operate similarly to an IRB, will review the ethics of conducting the proposed research. Other regulatory authorities around the rest of the world have slightly differing requirements involving both the execution of clinical studies and the import/export of pharmaceutical products. It is our responsibility to ensure we conduct our business in accordance with the regulations of each relevant territory.
In order to gain marketing approval, we must submit a dossier to the relevant authority for review, which is known in the U.S. as an NDA and in the E.U. as a marketing authorization application (“MAA”). The format is usually specific and laid out by each authority, although in general it will include information on the quality of the chemistry, manufacturing and pharmaceutical aspects of the product as well as the nonclinical and clinical data. Once the submitted NDA is accepted for filing by the FDA, it undertakes the review process that currently takes on average 10 months, unless an expedited priority review is granted which takes six months to complete. Approval can take several months to several years, if multiple 10-month review cycles are needed before final approval is obtained, if at all.
The approval process can be affected by a number of factors. The NDA may require additional preclinical, manufacturing data or clinical studies which may be requested at the end of the 10-month NDA review cycle, thereby delaying approval until additional data are submitted and may involve substantial unbudgeted costs.
In addition to obtaining approval for each product, in many cases each drug manufacturing facility must be approved. The regulatory authorities usually will conduct an inspection of relevant manufacturing facilities, and review manufacturing procedures, operating systems and personnel qualifications. Further inspections may occur over the life of the product. An inspection of the clinical investigation sites by a competent authority may be required as part of the regulatory approval procedure. As a condition of marketing approval, the regulatory agency may require post-marketing surveillance to monitor for adverse effects or other additional studies as deemed appropriate. After approval for the initial indication, further clinical studies may be necessary to gain approval for any additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect the marketability of a product.
The FDA offers a number of regulatory mechanisms that provide expedited or accelerated approval procedures for selected drugs in the indications on which we are focusing our efforts. These include accelerated approval under Subpart H of the agency’s NDA approval regulations, fast track drug development procedures, breakthrough drug designation and priority review. At this time, we have not determined whether any of these approval procedures will apply to our current drug candidates.
By leveraging existing preclinical and clinical safety and efficacy data, we seek to build upon an existing knowledge base to accelerate our research. In addition, through our focus on end-stage population which has no current treatment options, regulatory approval for commercialization may sometimes be achieved in an accelerated manner. Accelerated approval by the FDA in this category may be granted on objective response rates and duration of responses rather than demonstration of survival benefit. As a
26
result, studies of drugs to treat end-stage refractory cancer indications have historically involved fewer patients and generally have been faster to complete than studies of drugs for other indications. We are aware that the FDA and other similar agencies are regularly reviewing the use of objective endpoints for commercial approval and that policy changes may impact the size of studies required for approval, timelines and expenditures significantly.
The U.S., E.U. and other jurisdictions may grant orphan drug designation to drugs intended to treat a “rare disease or condition,” which, in the U.S., is generally a disease or condition that affects no more than 200,000 individuals. In the E.U., orphan drug designation can be granted if: the disease is life threatening or chronically debilitating and affects no more than 50 in 100,000 persons in the E.U.; without incentive, it is unlikely that the drug would generate sufficient return to justify the necessary investment; and no satisfactory method of treatment for the condition exists or, if it does, the new drug will provide a significant benefit to those affected by the condition. If a product that has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the U.S. and 10 years in the E.U. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for different indications. Orphan drug designation must be requested before submitting an NDA or MAA. After orphan drug designation is granted, the identity of the therapeutic agent and its potential orphan use are publicly disclosed. Orphan drug designation does not convey an advantage in, or shorten the duration of, the review and approval process. However, this designation provides an exemption from marketing and authorization fees charged to NDA sponsors under the Prescription Drug Act.
Maintaining substantial compliance with appropriate federal, state and local statutes and regulations requires the expenditure of substantial time and financial resources. Drug manufacturers are required to register their establishments with the FDA and certain state agencies, and after approval, the FDA and these state agencies conduct periodic unannounced inspections to ensure continued compliance with ongoing regulatory requirements, including cGMPs. In addition, after approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. The FDA may require post-approval testing and surveillance programs to monitor safety and the effectiveness of approved products that have been commercialized. Any drug products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including:
In addition, the FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. There are numerous regulations and policies that govern various means for disseminating information to health-care professionals as well as consumers, including to industry sponsored scientific and educational activities, information provided to the media and information provided over the Internet. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label.
The FDA has very broad enforcement authority and the failure to comply with applicable regulatory requirements can result in administrative or judicial sanctions being imposed on us or on the manufacturers and distributors of our approved products, including warning letters, refusals of government contracts, clinical holds, civil penalties, injunctions, restitution and disgorgement of profits, recall or seizure of products, total or partial suspension of production or distribution, withdrawal of approvals, refusal to approve pending applications, and criminal prosecution resulting in fines and incarceration. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. In addition, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market.
Sales of our product candidates, if approved, will depend, in part, on the extent to which such products will be covered by third-party payors, such as government health care programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly limiting coverage or reducing reimbursements for medical products and services. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price
27
controls, restrictions on reimbursement and requirements for substitution of generic products. Third-party payors decide which therapies they will pay for and establish reimbursement levels. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own coverage and reimbursement policies. However, decisions regarding the extent of coverage and amount of reimbursement to be provided for any drug candidates that we develop will be made on a payor-by-payor basis. Each payor determines whether or not it will provide coverage for a therapy, what amount it will pay the manufacturer for the therapy, and on what tier of its formulary it will be placed. The position on a payor’s list of covered drugs, or formulary, generally determines the co-payment that a patient will need to make to obtain the therapy and can strongly influence the adoption of such therapy by patients and physicians. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our product candidates or a decision by a third-party payor to not cover our product candidates could reduce physician usage of our product candidates, once approved, and have a material adverse effect on our sales, results of operations and financial condition.
Because of our current and future arrangements with healthcare professionals, principal investigators, consultants, customers and third-party payors, we will also be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we will conduct our business, including our clinical research, proposed sales, marketing and educational programs. Failure to comply with these laws, where applicable, can result in the imposition of significant civil penalties, criminal penalties, or both. The U.S. laws that may affect our ability to operate, among others, include: the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, which governs the conduct of certain electronic healthcare transactions and protects the security and privacy of protected health information; certain state laws governing the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; the federal healthcare programs’ Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent; federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; the Physician Payments Sunshine Act, which requires manufacturers of drugs, devices, biologics, and medical supplies to report annually to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and ownership and investment interests held by physicians and their immediate family members; and state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers.
In addition, many states have similar laws and regulations, such as anti-kickback and false claims laws that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs. Additionally, to the extent that our product is sold in a foreign country, we may be subject to similar foreign laws.
We are also subject to numerous environmental and safety laws and regulations, including those governing the use and disposal of hazardous materials. The cost of compliance with and any violation of these regulations could have a material adverse effect on our business and results of operations. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal regulations, accidental contamination or injury from these materials may occur. Compliance with laws and regulations relating to the protection of the environment has not had a material effect on our capital expenditures or our competitive position. However, we are not able to predict the extent of government regulation, and the cost and effect thereof on our competitive position, which might result from any legislative or administrative action pertaining to environmental or safety matters.
Competition
The development and commercialization of new drug products is highly competitive. We expect that we will face significant competition from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide with respect to VAL-083 and REM-001 and any other product candidates that we may seek to develop or commercialize in the future. Specifically, due to the large unmet medical need, global demographics and relatively attractive reimbursement dynamics, the oncology market is fiercely competitive and there are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of product candidates for the treatment of cancer. Our competitors may succeed in developing, acquiring or licensing technologies and drug products that are more effective, have fewer or more tolerable side effects or are less costly than any product candidates that we are currently developing or that we may develop, which could render our product candidates obsolete and noncompetitive.
28
All of the top ten global pharmaceutical companies, and many of the mid-size pharmaceutical companies, have a strong research and development and commercial presence in oncology. Smaller companies also focus on oncology, including companies such as Clovis Oncology, Inc., Epizyme, Inc., ImmunoGen, Inc., Incyte Corporation, Infinity Pharmaceuticals, Inc., MacroGenics, Inc., Merrimack Pharmaceuticals, Inc., Onconova Therapeutics, Inc., Pharmacyclics, Inc., Puma Biotechnology, Inc. and Seagen, Inc.
We are currently participating in the GCAR GBM AGILE Study to facilitate the advancement of VAL-083's clinical development. In addition to us, there are four product candidates being studied: Bayer Pharmaceuticals (Regorafenib), Kazia Therapeutics (Paxalisib), Biohaven Pharmaceutical Holding Company (troriluzole) and Vigeo Therapeutics (VT1021). All of these companies are seeking approval in GBM.
Several companies are marketing and developing oncology immunotherapy products. Companies with approved marketed oncology products for GBM are Merck (Temodar ®) and Genentech (Avastin ®). Companies with oncology immunotherapy product candidates in clinical development for GBM include, but are not limited to, Northwest Biotherapeutics (DCVax-L), Celldex Therapeutics (Rindopepimut (CDX-110) and ImmunoCellular Therapeutics (ICT-107).
We are not aware of any therapies specifically approved for CMBC in the US. IGEA Medical S.p.A. is developing an electro-chemotherapy treatment for CMBC. Pinnacle Biologics Inc., a subsidiary of Advanz Pharma Healthcare Corp., sells Photofrin, a first-generation PDT product for treatment of certain endobronchial non-small-cell lung cancers and esophageal cancers. Photofrin is currently in Phase 2 studies in recurrent glioma. To our knowledge, there is no reported development program for Photofrin in CMBC. Rogers Sciences Inc. is a medical device company that is developing a light delivery device for use with PDT treatment of cutaneous cancers that they are currently clinically testing in a Phase 2 study in CMBC patients.
There are numerous therapies currently used to treat CMBC patients including chemotherapy, radiation therapy, surgical excision, hyperthermia, cryotherapy, electro-chemotherapy, topical drugs and intra-lesional chemotherapy injections, but, to our knowledge, there are no PDT therapies currently approved by the FDA for the treatment of CMBC or similar cutaneous cancers. Some topical PDT agents have been approved by the FDA for actinic keratosis which is a precancerous skin condition and they have been approved in some other countries for some conditions that we believe pose low medical risk such as basal cell cancer and acne.
In the BCCNS field we are aware of approved drugs in the U.S., including vismodegib (Eviredge), Odomzo (sonidegib), imiquimod and topical fluorouracil that are sometimes use off-label. PellePharm also recently completed a Phase 3 study in BCCNS but, to our knowledge, has not received marketing approval.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other marketing approval for their products before we are able to obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of our existing and potential future competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical studies, obtaining marketing approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller, or early stage, companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical study sites and patient registration for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs.
We expect that our ability to compete effectively will depend upon our ability to:
Failure to do one or more of these activities could have an adverse effect on our business, financial condition or results of operations.
29
Corporate History
We are a Nevada corporation formed on June 24, 2009, under the name Berry Only Inc. On January 25, 2013, we entered into and closed an exchange agreement (the “Exchange Agreement”), with Del Mar (BC), Callco, Exchangeco and the security holders of Del Mar (BC). Upon completion of the Exchange Agreement, Del Mar (BC) became a wholly-owned subsidiary of ours (the “Reverse Acquisition”).
On August 19, 2020, we merged with Adgero and changed our name from DelMar Pharmaceuticals, Inc. to Kintara Therapeutics, Inc. We are the parent company to the following entities:
Research and Development
During the years ended June 30, 2022, and 2021, we recognized $15.2 million and $11.8 million, respectively, in research and development expenses.
Employees
We have three full-time employees and retain the services of approximately 20 persons on an independent contractor/consultant and contract-employment basis. As such, we currently operate in a “virtual” corporate structure in order to minimize fixed personnel costs.
Available Information
We maintain an internet website at www.kintara.com. We do not incorporate the information on our website into this report and you should not consider it part of this report.
30
Item 1A. Risk Factors
Summary of Risk Factors
An investment in our common stock involves a high degree of risk. In determining whether to purchase our common stock, an investor should carefully consider all of the material risks described below, together with the other information contained in this report before making a decision to purchase our securities. An investor should only purchase our securities if he or she can afford to suffer the loss of his or her entire investment.
Risks Related to Our Business
We have expressed substantial doubt about our ability to continue as a going concern.
As discussed in Note 1 to the consolidated financial statements for the year ended June 30, 2022, our consolidated financial statements for the year ended June 30, 2022, include an explanatory paragraph that such financial statements were prepared assuming that we will continue as a going concern. A going concern basis assumes that we will continue our operations for the foreseeable future and contemplates the realization of assets and the settlement of liabilities in the normal course of business.
We are in the clinical stage and have not generated any revenues to-date. For the year ended June 30, 2022, we reported a loss of approximately $22.7 million and a negative cash flow from operations of approximately $20.4 million. We had an accumulated deficit
31
of approximately $136.4 million and had cash and cash equivalents of approximately $11.8 million as of June 30, 2022. We do not have the prospect of achieving revenues until such time that our product candidates are commercialized, or partnered, which may not ever occur. In the near future, we will require additional funding to maintain our clinical studies, research and development projects, and for general operations. These circumstances indicate substantial doubt exists about our ability to continue as a going concern within one year from the date of filing of the consolidated financial statements.
Despite the completion of two registered direct financings during the year ended June 30, 2022 for aggregate net proceeds of approximately $21.6 million, we will need additional financing to complete all of our planned operations. Consequently, management is pursuing various financing alternatives to fund our operations so we can continue as a going concern. On August 2, 2022 we entered into a stock purchase agreement with Lincoln Park (the "Purchase Agreement"), pursuant to which Lincoln Park has committed to purchase up to $20.0 million of our shares of common stock, subject to certain limitations. However, due to the uncertainty of the timing and amount of cash available under the Purchase Agreement, the Purchase Agreement does not alleviate our going concern. In addition, the coronavirus (“COVID-19”) pandemic has created significant economic uncertainty and volatility in the credit and capital markets. Management plans to secure the necessary financing through the issue of new equity and/or the entering into of strategic partnership arrangements but the ultimate impact of the COVID-19 pandemic on our ability to raise additional capital is unknown and will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration of the COVID-19 outbreak and any new information which may emerge concerning the severity of the COVID-19 pandemic. We may not be able to raise sufficient additional capital and may tailor our drug candidates development programs based on the amount of funding we are able to raise in the future. Nevertheless, there is no assurance that these initiatives will be successful.
The financial statements do not give effect to any adjustments to the amounts and classification of assets and liabilities that may be necessary should we be unable to continue as a going concern. Such adjustments could be material.
We are a clinical stage company, have a history of operating losses, and expect to incur significant additional operating losses.
We are a clinical stage company with a history of operating losses. For the fiscal years ended June 30, 2022 and 2021, we had net losses of approximately $22.7 million and $38.3 million, respectively and an accumulated deficit of approximately $136.4 million at June 30, 2022. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in similar stages of operations. We expect to incur substantial additional net expenses and losses over the next several years as our research, development, clinical studies, and commercial activities increase.
The amount of future losses and when, if ever, we will achieve profitability are uncertain. Our ability to generate revenue and achieve profitability will depend on, among other things, successful completion of the preclinical and clinical development of our product candidates; obtaining necessary regulatory approvals from the FDA and international regulatory agencies; successful manufacturing, sales and marketing arrangements; and raising sufficient funds to finance our activities. If we are unsuccessful at some or all of these undertakings, our business, prospects and results of operations may be materially adversely affected.
We will need to raise additional capital, which may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to technologies or product candidates.
Until such time, if ever, as we can generate substantial product revenues, we expect to finance our cash needs through a combination of public or private equity offerings, debt financings and/or license and development agreements with collaboration partners. As of June 30, 2022, we had cash and cash equivalents of approximately $11.8 million. We expect the cash available at June 30, 2022, and the potential cash received from the sale of shares under our Purchase Agreement with Lincoln Park, to fund our planned operations for less than one year from the date of filing this report on Form 10-K. We will need to raise additional capital to fund our planned operations. While we have a Purchase Agreement with Lincoln Park, potential funds available under that agreement are not certain as to amount or timing and as a result, we do not have any committed external source of funds.
To the extent that we raise additional capital through sales of our common stock under our Lincoln Park agreement or through the sale of other equity or convertible debt securities, then-existing stockholders’ interests may be materially diluted, and the terms of such securities could include liquidation or other preferences that adversely affect their rights as common stockholders. Debt financing and preferred equity financing, if available, may involve agreements that include restrictive covenants that limit our ability to take specified actions, such as incurring additional debt, making capital expenditures or declaring dividends. In addition, debt financing would result in fixed payment obligations.
If we raise funds through collaborations, strategic partnerships or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves
32
Our inability to obtain additional financing could adversely affect our ability to meet our obligations under our planned clinical studies and could negatively impact the timing of our clinical results.
Our ability to meet our obligations and continue the research and development of our product candidates is dependent on our ability to continue to raise adequate financing. We may not be successful in obtaining such additional financing in the amount required at any time, or for any period, or, if available, that it can be obtained on terms satisfactory to us. In the event that we are unable to obtain such additional financing, we may be unable to meet our obligations under our planned clinical studies and we may have to tailor the drug development programs for our drug candidates based on the amount of funding we raise which could negatively impact the timing of our clinical results. In addition, we could be required to cease our operations.
We face significant risks related to the COVID-19 pandemic, or the widespread outbreak of any other communicable disease, which could have material and adverse impacts on our business, financial condition, liquidity and results of operations.
In December 2019 a novel strain of coronavirus, COVID-19 was reported to have surfaced in Wuhan, China and on March 11, 2020, it was declared a pandemic by the World Health Organization. The ultimate impact of the COVID-19 pandemic on our operations is unknown and will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration of the COVID-19 outbreak, new information which may emerge concerning the duration and severity of the COVID-19 pandemic, and any additional preventative and protective actions that governments, or us, may determine are needed.
The COVID-19 pandemic did not cause significant disruption to our Phase 2 clinical studies. Each of our now-completed Phase 2 clinical studies was conducted at respective single sites which reduced the risk of study disruption. Any disruptions to patient treatments for our Phase 2 studies were within allowances under each study protocol. Access to the sites by our clinical monitors was limited during the COVID-19 pandemic but the recording of study data in both studies and patient treatments at both study sites was conducted per protocol.
Regarding the VAL-083 study arm of the GBM AGILE Study that is currently being conducted at multiple sites in the United States, Canada, and Europe, we have not experienced any significant impacts on patient enrollment or treatment. With respect to the REM-001 drug supply, we are currently experiencing some delays in contract manufacturing schedules and supplies which we attribute to COVID-19. The current delays could have an impact on our REM-001 program timeline.
Changes in circumstances surrounding COVID-19, such as additional travel limitations imposed by governmental authorities could result in new patients being unable to be enrolled in our studies, or existing patients being unable to continue to receive treatment, could impact the cost of our studies as we may have to enroll additional patients in order to obtain the data necessary to be able to conclude our studies. We cannot predict whether any of our clinical testing sites will withdraw from participation in any of our studies temporarily, or permanently. In addition, even if we are able to fully enroll and treat all patients in our studies, obtaining full data could be impacted by an inability to ship and analyze samples, or otherwise complete data assessment. Further, if the patients enrolled in our clinical studies become infected with COVID-19, we may have more adverse events and deaths in our clinical studies as a result. We may also face difficulties enrolling patients in our clinical studies if the patient populations that are eligible for our clinical studies are impacted by the coronavirus disease. Vulnerable patients, such as the cancer patients enrolled in our clinical studies, may be at a higher risk of contracting COVID-19 and may experience more severe symptoms from the disease, adversely affecting our chances for regulatory approval, or requiring further clinical studies. The continued spread of COVID-19 globally, and the resulting travel restrictions in place by governments to help stop the spread of COVID-19, could adversely impact our clinical study operations, including the ability of our principal investigators and site staff to travel to our clinical study sites, and our ability to recruit and retain principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 if an outbreak occurs in their geography.
In addition, the continued impact resulting from the COVID-19 outbreak in areas where we have manufacturing operations for our clinical drug supply, or where our suppliers or distributors operate, or if the COVID-19 outbreak in these areas were to increase in severity, and the measures taken by the governments of countries affected, could adversely affect our business, financial condition, or results of operations by limiting our ability to manufacture or ship materials, or by forcing temporary closure of facilities that it relies upon.
The spread of COVID-19, which has caused a broad impact globally, may materially affect us economically. While the potential economic impact brought by, and the duration of, COVID-19 may be difficult to assess or predict, a widespread pandemic could result in significant disruption of global financial markets, reducing our ability to access capital, which could in the future negatively affect our liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect ongoing business and the value of our common stock.
33
While our common stock is expected to continue listing on The Nasdaq Capital Market LLC, there is no guarantee as to how long such listing will be maintained.
Our common stock is listed for trading on The Nasdaq Capital Market LLC. We must satisfy Nasdaq’s continued listing requirements, including, among other things, a minimum closing bid price requirement of $1.00 per share for 30 consecutive business days. If a company’s common stock trades for 30 consecutive business days below the $1.00 minimum closing bid price requirement, Nasdaq will send a deficiency notice, advising that such company has been afforded a “compliance period” of 180 calendar days to regain compliance with the applicable requirements. Thereafter, if such a company does not regain compliance with the bid price requirement, a second 180-day compliance period may be available, provided (i) it meets the continued listing requirement for market value of publicly held shares and all other applicable requirements for initial listing on Nasdaq, including stockholder equity requirements, which we may be unable to satisfy (except for the bid price requirement), and (ii) it provides written notice to Nasdaq of its intention to cure this deficiency during the second compliance period by effecting a reverse stock split, if necessary. In the event the company does not regain compliance with Rule 5550(a)(2) prior to the expiration of the initial 180 calendar day period, and if it appears to the Staff of the Listing Qualifications Department of The Nasdaq Stock Market LLC (the “Nasdaq Staff”) that the company will not be able to cure the deficiency, or if the company is not otherwise eligible, the Nasdaq Staff will provide the company with written notification that its securities are subject to delisting from Nasdaq. At that time, the company may appeal the delisting determination to a Hearings Panel.
On December 3, 2021, we received a letter from the Nasdaq Staff indicating that, based upon the closing bid price of our common stock for the prior 30 consecutive business days, we were not in compliance with the requirement to maintain a minimum bid price of $1.00 per share for continued listing on Nasdaq, as set forth in Nasdaq Listing Rule 5550(a)(2) (the “Minimum Bid Price Requirement”). In accordance with Nasdaq Listing Rule 5810(c)(3)(A), we were provided a grace period of 180 days, or until June 1, 2022, to regain compliance with the Minimum Bid Price Requirement.
On May 17, 2022, we submitted a request to Nasdaq for an additional 180-day extension to regain compliance with the Minimum Bid Price Requirement. On June 2, 2022, we received a letter from Nasdaq advising that we had been granted a 180-day extension to November 28, 2022, to regain compliance with the Minimum Bid Price Requirement, in accordance with Nasdaq Listing Rule 5810(c)(3)(A).
We will continue to monitor the closing bid price of our common stock and may, if appropriate, consider implementing available options, including but not limited to, implementing a reverse stock split of our outstanding securities, to regain compliance with the Minimum Bid Price Requirement. If we do not regain compliance within the allotted compliance period, Nasdaq will provide notice that our common stock will be subject to delisting. We would then be entitled to appeal that determination to a Nasdaq hearings panel. There can be no assurance that we will regain compliance with the Minimum Bid Price Requirement during this 180-day extension.
If we fail to meet any of the other continued listing requirements, including stockholder equity requirements, our securities may be delisted from The Nasdaq Capital Market LLC and trade on the OTC Markets Group Inc. or other small trading markets, which could reduce the liquidity of our common stock materially and result in a corresponding material reduction in the price of our common stock. In addition, delisting could harm our ability to raise capital through alternative financing sources on terms acceptable to us, or at all, and may result in the potential loss of confidence by investors, employees and business development opportunities. Such a delisting likely would impair your ability to sell or purchase our common stock when you wish to do so. Further, if we were to be delisted from Nasdaq, our common stock may no longer be recognized as a “covered security” and we would be subject to regulation in each state in which it offers securities. Thus, delisting from Nasdaq could adversely affect our ability to raise additional financing through the public or private sale of equity securities, would significantly impact the ability of investors to trade our securities and would negatively impact the value and liquidity of our common stock.
The Series C Preferred Stock has rights, preferences and privileges that will not be held by, and will be preferential to, the rights of holders of our common stock, which could adversely affect the liquidity and financial condition of the Company, and may result in the interests of the holders of Series C Preferred Stock differing from those of the holders of our common stock.
The Series C Preferred Stock ranks on parity with the shares of our Series A Preferred Stock with respect to liquidation preferences. Upon any dissolution, liquidation or winding up, whether voluntary or involuntary, holders of Series C Preferred Stock will be entitled to receive distributions out of our assets in an amount per share equal to $1,000 plus all accrued and unpaid dividends, whether capital or surplus before any distributions shall be made on any shares of our common stock.
In addition, holders of Series C Preferred Stock will be entitled to dividends, payable in shares of our common stock at a rate of 10%, 15%, 20% and 25% of the number of shares of common stock issuable upon conversion of the Series C Preferred Stock, on the 12th, 24th, 36th and 48th month, anniversary of the initial closing of the Private Placement, which occurred on August 19, 2020.
34
Dividends will be payable in shares of our common stock and will only be payable to those holders that continue to hold Series C Preferred Stock on the respective anniversary dates of August 19, 2020.
These dividend obligations to the holders of Series C Preferred Stock could limit our ability to obtain additional financing, which could have an adverse effect on our financial condition. The preferential rights described above could also result in divergent interests between the holders of shares of Series C Preferred Stock and the holders of our common stock.
Any issuance of our common stock upon conversion of the Series C Preferred Stock will cause dilution to our then existing stockholders and may depress the market price of our common stock.
The Series C Preferred Stock accrues dividends in shares of our common stock at an initial rate of 10% per annum and following the forty-eight-month anniversary of the initial closing of the Private Placement which occurred on August 19, 2020, such dividend rate will increase to 25% per annum. Each class of Series C Preferred Stock has a Conversion Price that will be equal to the lesser of (i) the closing price of our common stock on Nasdaq on the date immediately preceding the signing of the applicable binding agreements for the applicable closing date of the Private Placement for which the Series C Preferred Stock is issued or (ii) the average closing price of the our common stock on Nasdaq for the five trading days immediately preceding the signing of the applicable binding agreements for the applicable closing date of the Private Placement for which the Series C Preferred Stock is issued, subject to adjustment. The Conversion Prices for the Series C-1 Preferred Stock, Series C-2 Preferred Stock and Series C-3 Preferred Stock are $1.16, $1.214 and $1.15, respectively.
The issuance of our common stock upon conversion of the Series C Preferred Stock and as payment of dividends on the Series C Preferred Stock will result in immediate and substantial dilution to the interests of holders of our common stock, and such dilution will increase over time in connection with the accrual of dividends on the Series C Preferred Stock.
We may incur future indebtedness that will rank senior to the Series C Preferred Stock or issue additional series of preferred stock that rank on a parity with, or senior to, the Series C Preferred Stock as to dividend payments and liquidation preference.
We may incur substantial amounts of additional debt and other obligations that will rank senior to the Series C Preferred Stock, and the terms of the Series C Preferred Stock do not limit the amount of such debt or other obligations that we may incur. The terms of the Series C Preferred Stock will not prohibit us from issuing additional series of preferred stock that would rank on parity with the Series C Preferred Stock. The Articles allow for the board of directors to create new series of preferred stock without further approval by its stockholders, which could adversely affect the rights of the holders of the Series C Preferred Stock and common stock. The issuances of other series of preferred stock could have the effect of reducing the amounts available to the Series C Preferred Stock in the event of liquidation. If we issue preferred stock with voting rights that dilute the voting power of our common stock, the market price of our common stock could decrease, adversely affecting the value of the Series C Preferred Stock. Additional issuances and sales of preferred stock, or the perception that such issuances and sales could occur, may cause prevailing market prices for our common stock to decline and may adversely affect our ability to raise additional capital in the financial markets at times and prices favorable to it.
If we are unable to effectively maintain a system of internal control over financial reporting, we may not be able to accurately or timely report our financial results and our stock price could be adversely affected.
Section 404 of the Sarbanes-Oxley Act of 2002 and related regulations require us to evaluate the effectiveness of our internal control over financial reporting as of the end of each fiscal year, and to include a management report assessing the effectiveness of our internal control over financial reporting in our Annual Report on Form 10-K for that fiscal year. Any failure to implement new or improved controls, or difficulties encountered in the implementation or operation of these controls, could harm our operations, decrease the reliability of our financial reporting, and cause us to fail to meet our financial reporting obligations, which could adversely affect our business and reduce our stock price.
We are subject to state laws in California that require gender and diversity quotas for boards of directors of public companies headquartered in California.
In September 2018, California enacted SB 826, requiring public companies headquartered in California to maintain minimum female representation on their boards of directors as follows: by December 31, 2019, public company boards must have a minimum of one female director; by December 31, 2021, public company boards with five members will be required to have at least two female directors, and public company boards with six or more members will be required to have at least three female directors. On May 13, 2022, the Los Angeles Superior Court declared SB 826 unconstitutional and, although the California Secretary of State has directed counsel to file an appeal of the decision, the State of California is currently precluded from enforcing SB 826.
35
Additionally, on September 30, 2020, California enacted AB 979, requiring public companies with principal executive offices in California to each have at least one director from an underrepresented community based on ethnicity and sexual orientation by December 31, 2021. By December 31, 2022, each of these companies will be required to have at least two directors from such underrepresented communities if such company has more than four but fewer than nine directors, or at least three directors from underrepresented communities if the company has nine or more directors. On April 1, 2022, the Los Angeles Superior Court declared AB 979 unconstitutional and, although the California Secretary of State has filed a notice of appeal in the case, the State of California is currently precluded from enforcing AB 979.
If the State of California successfully appeals the court decisions regarding SB 826 or AB 979, we cannot assure that we can recruit, attract and/or retain qualified members of the board and continue to meet gender and diversity quotas as required by California law (provided that such laws are not repealed before the compliance deadlines), which may cause certain investors to divert their holdings in our securities and expose us to financial penalties and/or reputational harm.
We are a clinical stage company and may never achieve commercialization of our product candidates or profitability.
We are a clinical stage of development and commercialization of our technologies and product candidates. We have not yet begun to market any products and, accordingly, have not begun or generate revenues from the commercialization of our products. Our products will require significant additional clinical testing and investment prior to commercialization. A commitment of substantial resources by us and, potentially, our partners to conduct time-consuming research and clinical studies will be required if we are to complete the development of our product candidates. There can be no assurance that our product candidates will meet applicable regulatory standards, obtain required regulatory approvals, be capable of being produced in commercial quantities at reasonable costs or be successfully marketed. Our product candidates are not expected to be commercially available for several years, if at all.
We are currently focused on the development of two product candidates.
Our product development efforts are currently focused on two product candidates: VAL-083 for GBM and REM-001 for CMBC. If either VAL-083 or REM-001 fail to achieve clinical endpoints or exhibit unanticipated toxicity or if a superior product is developed by a competitor, our prospects for obtaining regulatory approval and commercialization for either candidate may be negatively impacted. In the long-term, we hope to establish a pipeline of multiple product candidates. However, at this time we do not have any formal agreements granting us any rights to such additional product candidates.
Even if we are able to commercialize any product candidate that we develop, the product may become subject to unfavorable pricing regulations, third-party payor reimbursement practices or healthcare reform initiatives that could harm our business.
The commercial success of our current or future product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of our product candidates will be paid by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities (such as Medicare and Medicaid), private health coverage insurers and other third-party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our products. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish and maintain pricing sufficient to realize a meaningful return on our investment.
There is significant uncertainty related to third-party payor coverage and reimbursement of newly approved drugs. Marketing approvals, pricing and reimbursement for new drug products vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some non-U.S. markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that delay commercial launch of the product, possibly for lengthy time periods, which may negatively impact the revenues we are able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more product candidates, even if our product candidates obtain marketing approval.
Our ability to commercialize VAL-083, REM-001, or any other product candidates will depend in part on the extent to which coverage and reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will cover and establish reimbursement levels. The healthcare industry is acutely focused on cost containment, both in the United States and elsewhere. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications, which could affect our ability to sell our product candidates profitably. These payors may not view our products, if any, as cost-effective, and coverage and reimbursement may not be available to our customers, or may not be sufficient to allow our products, if any, to be
36
marketed on a competitive basis. Cost-control initiatives could cause us to decrease the price we might establish for products, which could result in lower than anticipated product revenues. If the prices for our products, if any, decrease or if governmental and other third-party payors do not provide adequate coverage or reimbursement, our prospects for revenue and profitability will suffer.
There may also be delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the indications for which the drug is approved by the FDA or comparable non- U.S. regulatory authorities. Moreover, eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Reimbursement rates may vary, by way of example, according to the use of the drug and the clinical setting in which it is used. Reimbursement rates may also be based on reimbursement levels already set for lower cost drugs or may be incorporated into existing payments for other services.
In addition, increasingly, third-party payors are requiring higher levels of evidence of the benefits and clinical outcomes of new technologies and are challenging the prices charged. We cannot be sure that coverage will be available for any product candidate that we, or third-parties, commercialize and, if available, that the reimbursement rates will be adequate. Further, the net reimbursement for drug products may be subject to additional reductions if there are changes to laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. An inability to promptly obtain coverage and adequate payment rates from both government-funded and private payors for any of our product candidates for which we obtain marketing approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Pursuant to the terms of our Series B Preferred Stock that has now been fully converted to common stock, the Valent Technologies, LLC (“Valent”) Patent Assignment Agreement, and the St. Cloud Agreement we may be required to pay royalties.
Pursuant to the terms of the Valent Patent Assignment Agreement, as amended, and our Series B Preferred Stock Certificate of Designation and the related Series B Preferred Royalty Agreement, we will be required to pay royalties if we receive revenue from product sales or from the partnering of VAL-083. If we obtain FDA, EMA, or other regulatory approvals of VAL-083, and/or if we generate sales of VAL-083, or we receive any proceeds from the licensing or other disposition of VAL-083, we are required to pay to the former holders of our Series B Preferred Stock, a single-digit royalty. In addition, we are required to pay a future royalty on all revenues derived from the development and commercialization of VAL-083 to Valent. The royalty payment rights will expire when the patents covering the applicable product expire.
Also, under our St. Cloud agreement, we must pay to St. Cloud and Steven Rychnovsky, PhD, in the aggregate, a royalty fee of six percent (6%) of net sales during the royalty term on a country-by-country and product-by-product basis with St. Cloud receiving a royalty rate of four and eight tenths percent (4.8%) and Steven Rychnovsky, PhD, receiving a royalty of one and two tenths percent (1.2%). The royalty term for a product commences on the first commercial sale of the product, such as REM-001 Therapy, in any country, and the royalty fee must be paid within 30 days of each calendar quarter during which revenue is collected. The royalty term terminates on the later of (i) the invalidation, revocation, lapse or expiration of the last to expire valid claim on any patent acquired in the St. Cloud Agreement that would be infringed by the sale of the product in the country where the commercial sale takes place or (ii) the expiration of the period for which we hold exclusive marketing rights of the product in the country, if we are granted those rights under the St. Cloud Agreement.
We are dependent on obtaining certain patents and protecting our proprietary rights.
Our success will depend, in part, on our ability to obtain patents, maintain trade secret protection and operate without infringing on the proprietary rights of third parties or having third parties circumvent our rights. We have filed and are actively pursuing patent applications for our products. The patent positions of biotechnology, biopharmaceutical and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions. Thus, there can be no assurance that any of our patent applications will result in the issuance of patents, that we will develop additional proprietary products that are patentable, that any patents issued to us or those that already have been issued will provide us with any competitive advantages or will not be challenged by any third parties, that the patents of others will not impede our ability to do business or that third parties will not be able to circumvent our patents. Furthermore, there can be no assurance that others will not independently develop similar products, duplicate any of our products not under patent protection, or, if patents are issued to us, design around the patented products we developed or will develop.
We may be required to obtain licenses from third parties to avoid infringing patents or other proprietary rights. No assurance can be given that any licenses required under any such patents or proprietary rights would be made available, if at all, on terms we find acceptable. If we do not obtain such licenses, we could encounter delays in the introduction of products or could find that the development, manufacture or sale of products requiring such licenses could be prohibited.
37
A number of pharmaceutical, biopharmaceutical and biotechnology companies and research and academic institutions have developed technologies, filed patent applications or received patents on various technologies that may be related to or affect our business. Some of these technologies, applications or patents may conflict with our technologies or patent applications. Such conflict could limit the scope of the patents, if any, that we may be able to obtain or result in the denial of our patent applications. In addition, if patents that cover our activities are issued to other companies, there can be no assurance that we would be able to obtain licenses to these patents at a reasonable cost or be able to develop or obtain alternative technology. If we do not obtain such licenses, we could encounter delays in the introduction of products, or could find that the development, manufacture or sale of products requiring such licenses could be prohibited. In addition, we could incur substantial costs in defending ourselves in suits brought against us on patents it might infringe or in filing suits against others to have such patents declared invalid.
Patent applications in the U.S. are maintained in secrecy and not published if either: (i) the application is a provisional application or (ii) the application is filed and we request no publication, and certify that the invention disclosed “has not and will not” be the subject of a published foreign application. Otherwise, U.S. applications or foreign counterparts, if any, publish 18 months after the priority application has been filed. Since publication of discoveries in the scientific or patent literature often lag behind actual discoveries, we cannot be certain that we or any licensor were the first creator of inventions covered by pending patent applications or that we or such licensor was the first to file patent applications for such inventions. Moreover, we might have to participate in interference proceedings declared by the U.S. Patent and Trademark Office (the “USPTO”) to determine priority of invention, which could result in substantial cost to us, even if the eventual outcome were favorable to us. There can be no assurance that our patents, if issued, would be held valid or enforceable by a court or that a competitor’s technology or product would be found to infringe such patents.
Moreover, we may be subject to third-party preissuance submissions of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights.
Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us, or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our patents by developing similar or alternative technologies or products in a non-infringing manner.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our patents may be challenged in courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate, or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
In addition, the protection of intellectual property rights in China (where one of our clinical product candidates, VAL-083, is manufactured pursuant to a collaboration agreement with the only manufacturer presently licensed by the CFDA to manufacture VAL-083 for the China market, and where VAL-083 is approved for the treatment of CML and lung cancer) is relatively weak compared to the United States, which may negatively affect our ability to generate royalty revenue from sales of VAL-083 in China.
Much of our know-how and technology may not be patentable. To protect our rights, we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. There can be no assurance, however, that these agreements will provide meaningful protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure. Further, our business may be adversely affected by competitors who independently develop competing technologies, especially if we obtain no, or only narrow, patent protection.
We do not hold any patents covering our laser light source or light delivery device for REM-001.
Our laser light source and light delivery device are not currently covered by any patents; We do not have any patents pending, and do not currently intend to seek patent protection for these devices. As a result, competitors may be able to offer and sell products or drug delivery technology, as the case may be, using the same technology as our laser light source and/or light delivery devices, so long as these competitors do not infringe any other valid patents that it or third-parties hold.
38
While we plan to protect our proprietary information related to our laser light source and light delivery device as trade secrets through certain agreements with our employees, consultants, agents and other organizations to which we have disclosed our proprietary information, we cannot give any assurance that these agreements will provide effective protection for our proprietary information in the event of unauthorized use or disclosure of such information. If other laser light sources or light delivery devices are approved and marketed, we will be unable to prevent them from competing with REM-001 Therapy in the marketplace using a different drug molecule that is not encompassed by any of our owned or licensed patents. We expect that the presence of one or more competing products would reduce our market share and could negatively impact price levels and third-party reimbursement policies for REM-001 Therapy, any of which would materially affect our business.
We may be unable to protect our patents and proprietary rights.
Our future success will depend to a significant extent on our ability to:
We can provide no assurance that our patent rights will afford any competitive advantages and these rights may be challenged or circumvented by third parties. Further, patents may not be issued on any of our pending patent applications in the U.S. or abroad. Because of the extensive time required for development, testing and regulatory review of a product candidate, it is possible that before a product candidate can be commercialized, any related patent may expire, or remain in existence for only a short period following commercialization, reducing or eliminating any advantage of the patent.
If we sue others for infringing on our patents, a court may determine that such patents are invalid or unenforceable. Even if the validity of our patent rights is upheld by a court, a court may not prevent the alleged infringement of our patent rights on the grounds that such activity is not covered by our patent claims.
In addition, third parties may sue us for infringing on their patents. In the event of a successful claim of infringement against us, we may be required to:
If required, we can provide no assurance that we will be able to obtain such licenses on acceptable terms, or at all. If we are sued for infringement, we could encounter substantial delays in development, manufacture and commercialization of our product candidates. Any litigation, whether to enforce our patent rights or to defend against allegations that we infringed third-party rights, will be costly, time consuming, and may distract management from other important tasks.
As is commonplace in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. To the extent our employees are involved in research areas which are similar to those areas in which they were involved at their former employers, we may be subject to claims that such employees and/or we have inadvertently or otherwise used or disclosed the alleged trade secrets or other proprietary information of the former employers. Litigation may be necessary to defend against such claims, which could result in substantial costs and be a distraction to management and which may have a material adverse effect on us, even if we are successful in defending such claims.
39
We are subject to various government regulations.
The manufacture and sale of human therapeutic and diagnostic products in the U.S., Canada and foreign jurisdictions are governed by a variety of statutes and regulations. These laws require approval of manufacturing facilities, controlled research and testing of products and government review and approval of a submission containing manufacturing, preclinical and clinical data in order to obtain marketing approval based on establishing the safety and efficacy of the product for each use sought, including adherence to current cGMP during production and storage, and control of marketing activities, including advertising and labeling.
VAL-083, REM-001 and any other products we may develop will require significant development, preclinical and clinical testing and investment of substantial funds prior to its commercialization. The process of obtaining required approvals can be costly and time-consuming, and there can be no assurance that we will successfully develop any future products that will prove to be safe and effective in clinical studies or receive applicable regulatory approvals. Markets other than the U.S. and Canada have similar restrictions. Potential investors and shareholders should be aware of the risks, problems, delays, expenses and difficulties which we may encounter in view of the extensive regulatory environment which controls our business.
We may request priority review for our product candidates in the future. The FDA may not grant priority review for our product candidates. Moreover, even if the FDA designated such product for priority review, that designation may not lead to a faster regulatory review or approval process and, in any event, would not assure FDA approval.
We may be eligible for priority review designation for our product candidates if the FDA determines such product candidate offers major advances in treatment or provides a treatment where no adequate therapy exists. A priority review designation means that the goal for the FDA to review an application is six months, rather than the standard review period of ten months. The FDA has broad discretion with respect to whether or not to grant priority review status to a product candidate, so even if we believe a particular product candidate is eligible for such designation or status, the FDA may decide not to grant it. Thus, while the FDA has granted priority review to other oncology disease products, our product candidates, should we determine to seek priority review, may not receive similar designation. Moreover, even if our product candidate is designated for priority review, such a designation does not necessarily mean a faster regulatory review process or necessarily confer any advantage with respect to approval compared to conventional FDA procedures. Receiving priority review from the FDA does not guarantee approval within an accelerated timeline or thereafter.
We believe we may in some instances be able to secure approval from the FDA or comparable non-U.S. regulatory authorities to use accelerated development pathways. If we are unable to obtain such approval, we may be required to conduct additional preclinical studies or clinical studies beyond those that it contemplates, which could increase the expense of obtaining, and delay the receipt of, necessary marketing approvals.
We anticipate that we may seek an accelerated approval pathway for our product candidates. Under the accelerated approval provisions in the Federal Food, Drug, and Cosmetic Act (“FDCA”), and the FDA’s implementing regulations, the FDA may grant accelerated approval to a product designed to treat a serious or life-threatening condition that provides meaningful therapeutic benefit over available therapies upon a determination that the product has an effect on a surrogate endpoint or intermediate clinical endpoint that is reasonably likely to predict clinical benefit. The FDA considers a clinical benefit to be a positive therapeutic effect that is clinically meaningful in the context of a given disease, such as irreversible morbidity or mortality. For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. An intermediate clinical endpoint is a clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit. The accelerated approval pathway may be used in cases in which the advantage of a new drug over available therapy may not be a direct therapeutic advantage, but is a clinically important improvement from a patient and public health perspective. If granted, accelerated approval is usually contingent on the sponsor’s agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the drug’s clinical benefit. If such post-approval studies fail to confirm the drug’s clinical benefit, the FDA may withdraw its approval of the drug.
Prior to seeking such accelerated approval, we will seek feedback from the FDA and will otherwise evaluate our ability to seek and receive such accelerated approval. There can also be no assurance that after our evaluation of the feedback and other factors we will decide to pursue or submit an NDA, for accelerated approval or any other form of expedited development, review or approval. Similarly, there can be no assurance that after subsequent FDA feedback that we will continue to pursue or apply for accelerated approval or any other form of expedited development, review or approval, even if we initially decide to do so. Furthermore, if we decide to submit an application for accelerated approval or under another expedited regulatory designation (e.g., breakthrough therapy designation), there can be no assurance that such submission or application will be accepted or that any expedited development, review or approval will be granted on a timely basis, or at all. The FDA or other non-U.S. authorities could also require us to conduct further studies prior to considering our application or granting approval of any type. A failure to obtain accelerated approval or any
40
other form of expedited development, review or approval for any of our product candidates that we decide to seek accelerated approval for would result in a longer time period to commercialization of such product candidate, could increase the cost of development of such product candidate and could harm our competitive position in the marketplace.
We have conducted, and may in the future conduct, clinical studies for certain of our product candidates at sites outside the United States, and the FDA may not accept data from studies conducted in such locations.
We have conducted and may in the future choose to conduct one or more of our clinical studies outside the United States. Although the FDA may accept data from clinical studies conducted outside the United States, acceptance of this data is subject to certain conditions imposed by the FDA. For example, the clinical study must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The study population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. Generally, the patient population for any clinical studies conducted outside of the United States must be representative of the population for whom we intend to seek approval in the United States. In addition, while these clinical studies are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that the studies also complied with all applicable U.S. laws and regulations. There can be no assurance that the FDA will accept data from studies conducted outside of the United States. If the FDA does not accept the data from any of our clinical studies that we determine to conduct outside the United States, it would likely result in the need for additional studies, which would be costly and time-consuming and delay or permanently halt our development of the product candidate.
In addition, the conduct of clinical studies outside the United States could have a significant impact on us. The risks inherent in conducting international clinical studies include:
If our clinical studies fail to demonstrate safety and efficacy to the satisfaction of the FDA and comparable non-U.S. regulators, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
We are not permitted to commercialize, market, promote or sell any product candidate in the United States without obtaining marketing approval from the FDA. Comparable non-U.S. regulatory authorities, such as the EMA, impose similar restrictions. We may never receive such approvals. We must complete extensive preclinical development and clinical studies to demonstrate the safety and efficacy of our product candidates in humans before we will be able to obtain these approvals.
Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. We have not previously submitted an NDA to the FDA or similar drug approval filings to comparable non-U.S. regulatory authorities for any product candidate.
Any inability to successfully complete preclinical and clinical development could result in additional costs to us and impair our ability to generate revenues from product sales, regulatory and commercialization milestones and royalties. In addition, if (1) we are required to conduct additional clinical studies or other testing of our product candidates beyond the studies and testing that we contemplate, (2) we are unable to successfully complete clinical studies of our product candidates or other testing, (3) the results of these studies or tests are unfavorable, uncertain or are only modestly favorable, or (4) there are unacceptable safety concerns associated with our product candidate, we, in addition to incurring additional costs, may:
41
If we experience any of a number of possible unforeseen events in connection with clinical studies of our product candidates, potential marketing approval or commercialization of our product candidates could be delayed or prevented.
We may experience numerous unforeseen events during, or as a result of, clinical studies that could delay or prevent marketing approval of our product candidates, including:
42
Product development costs for us will increase if we experience delays in testing or pursuing marketing approvals and we may be required to obtain additional funds to complete clinical studies and prepare for possible commercialization of our product candidates. We do not know whether any preclinical tests or clinical studies will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant preclinical or clinical study delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates and may harm our business and results of operations. In addition, many of the factors that cause, or lead to, clinical study delays may ultimately lead to the denial of marketing approval of our product candidates.
If we experience delays or difficulties in the enrollment of patients in clinical studies, our product candidates may not achieve clinical development on our anticipated timeline, or at all, and our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical studies for VAL-083, REM-001 or any other product candidate if we are unable to locate and enroll a sufficient number of eligible patients to participate in clinical studies. Patient enrollment is a significant factor in the timing of clinical studies, and is affected by many factors, including:
Our inability to enroll a sufficient number of patients for our clinical studies could result in significant delays or may require us to abandon one or more clinical studies altogether. Enrollment delays in our clinical studies may result in increased development costs for our product candidates, delay or halt the development of and approval processes for our product candidates and jeopardize our ability to achieve our clinical development timeline and goals, including the dates by which we will commence, complete and receive results from clinical studies. Enrollment delays may also delay or jeopardize our ability to commence sales and generate revenues from our product candidates. Any of the foregoing could cause our value to decline and limit our ability to obtain additional financing, if needed.
Positive results in previous clinical studies of VAL-083 and REM-001 may not be replicated in future clinical studies, which could result in development delays or a failure to obtain marketing approval.
Positive results in previous clinical studies of VAL-083 and REM-001 may not be predictive of similar results in future clinical studies. Also, interim results during a clinical study do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical studies even after achieving promising results in early-stage development. Accordingly, the results from the completed preclinical studies and clinical studies for VAL-083 and REM-001 may not be predictive of the results we may obtain in later stage studies. Our clinical studies may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical studies. Moreover, clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical studies have nonetheless failed to obtain FDA or EMA, or other regulatory agency, approval for their products.
If we are required by the FDA to obtain approval of a companion diagnostic test in connection with approval of VAL-083, and we do not obtain, or face delays in obtaining such FDA approval, we may not be able to commercialize VAL-083 and our ability to generate revenue will be materially impaired.
If the FDA determines that a companion diagnostic device is necessary to the safe and effective use of a novel therapeutic product, the FDA will require that the companion diagnostic be approved or cleared for that indication along with that therapeutic
43
product. Companion diagnostics are developed in conjunction with clinical programs for the associated therapeutic product candidate and are subject to regulation as medical devices by the FDA.
If the FDA requires a companion diagnostic for the safe and effective use of VAL-083 in the MGMT unmethylated GBM population and a satisfactory companion diagnostic is not approved and commercially available, we may be required to create or obtain one that would be subject to regulatory approval requirements. The process of obtaining or creating such diagnostic is time consuming and costly. Any delay or failure to develop or obtain regulatory approval or clearance of a companion diagnostic could delay or prevent approval or continued marketing of such drug product candidate.
FDA approval of VAL-083, REM-001, or future product candidates may be denied.
There can be no assurance that the FDA will ultimately approve our NDAs. The FDA may deny approval of VAL-083 or REM-001 for many reasons, including:
If VAL-083 or REM-001 fail to receive FDA approval, our business and prospects will be materially adversely impacted.
We expect to rely on orphan drug status to develop and commercialize our product candidates, but our orphan drug designations may not confer marketing exclusivity or other expected commercial benefits as anticipated.
Market exclusivity afforded by orphan drug designation is generally offered as an incentive to drug developers to invest in developing and commercializing products for unique diseases that impact a limited number of patients. The FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. Qualification to maintain orphan drug status is generally monitored by the regulatory authorities during the orphan drug exclusivity period, currently seven years from the date of approval in the United States.
For VAL-083, we have been granted orphan drug designation in the United States for GBM, ovarian cancer, and medulloblastoma, and in Europe for GBM. In addition, for REM-001 the FDA granted our request that tin ethyl etiopurpurin (the active pharmaceutical ingredient in REM-001) be designated as an orphan drug for treatment of BCCNS. We also hold an orphan drug designation that was initially awarded to Miravant for tin ethyl etiopurpurin for the prevention of access graft disease in hemodialysis patients.
We expect to rely on orphan drug exclusivity for our product candidates. It is possible that the incidence and prevalence numbers for GBM, CMBC, and access graft disease could change. Should the incidence and prevalence of these diseases materially increase, it is possible that the orphan drug designation, and related market exclusivity, in the United States could be lost. Further, while we have been granted these orphan designations, the FDA can still approve different drugs for use in treating the same indication or disease, which would create a more competitive market for us and our revenues, if any, will be diminished.
Further, it is possible that another company also holding orphan drug designation for the same product candidate will receive marketing approval for the same indication before we do. If that were to happen, our applications for that indication may not be approved until the competing company’s period of exclusivity expires. Even if we are the first to obtain marketing authorization for an orphan drug indication, there are circumstances under which a competing product may be approved for the same indication during the seven-year period of marketing exclusivity, such as if the later product is shown to be clinically superior to the orphan product, or if the later product is deemed a different product than ours. Further, the seven-year marketing exclusivity would not prevent competitors from obtaining approval of the same product candidate as ours for indications other than those in which we have been granted orphan drug designation, or for the use of other types of products in the same indications as our orphan products.
44
If the market opportunities for our product candidates are smaller than we believe they are, our future revenues may be adversely affected and our business may suffer. Because the target patient populations of our product candidates are small, we must be able to successfully identify patients and capture a significant market share to achieve and maintain profitability.
We focus our research and product development on treatments for orphan cancer indications. Our projections of both the number of people who have failed other therapies or have limited medical options, are based on estimates. These estimates may prove to be incorrect and new studies may change the estimated incidence or prevalence. The number of patients in the United States, Europe and elsewhere may turn out to be lower than expected or may not be otherwise amenable to treatment with our products, or new patients may become increasingly difficult to identify or gain access to, all of which would adversely affect our results of operations and our business. Additionally, because our target patient populations are small, we will be required to capture a significant market share to achieve and maintain profitability.
We may be required to suspend or discontinue clinical studies due to unexpected side effects or other safety risks that could preclude approval of our products.
Our clinical studies may be suspended at any time for a number of reasons. For example, we may voluntarily suspend or terminate our clinical studies if at any time we believe that they present an unacceptable risk to the clinical study patients. In addition, the FDA or other regulatory agencies may order the temporary or permanent discontinuation of our clinical studies at any time if they believe that the clinical studies are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical study patients.
Administering any product candidate to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical studies of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidates for any or all targeted indications. Ultimately, some or all of our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects or even death as a result of participating in our clinical studies.
Our product candidates may cause undesirable adverse events or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, and/or result in significant negative consequences following regulatory approval, if any, including withdrawal from the market.
The REM-001 Therapy may exhibit undesirable and unintended side effects that may prevent or limit its commercial adoption and use. Even upon receiving approval by the FDA and other regulatory authorities, our products may later exhibit adverse side effects that prevent widespread use or necessitate withdrawal from the market. The manifestation of such side effects could cause its business to suffer.
Undesirable adverse events caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical studies and may result in a more restrictive label, a delay or denial of regulatory approval by the FDA or other comparable regulatory authorities, or a significant change in our clinical protocol or even our development plan. For example, in the four clinical studies of REM-001 therapy conducted by Miravant, there were a total of 17 serious adverse events, a large portion of which were related to necrosis of treated lesions. One adverse event that has been seen with REM-001 Therapy is a period of photosensitivity after receiving REM-001 Therapy. This period of photosensitivity is generally dose dependent and typically declines over time. A second such adverse event is pain that arises or results from the treatment. Treatment-related pain has been experienced by some patients and it is often treated with analgesics but in some cases more aggressive treatment can be required.
If clinical studies of our product candidates reveal a high and unacceptable severity or prevalence of certain adverse events, our studies could be suspended or terminated and the FDA and/or other comparable regulatory authorities could order us to cease further development of, or deny approval of, our product candidates for any or all targeted indications. Adverse events related to our candidates also may affect patient recruitment or the ability of enrolled subjects to complete the study and could result in potential liability claims. Any of these occurrences may significantly harm our reputation, business, financial condition and prospects.
Additionally, adverse events associated with our future approved products candidates may lead to potentially significant negative consequences, which include, but are not limited to, the following:
45
Any of these events could prevent us from achieving or maintaining market acceptance of any particular product candidate that is approved and could significantly harm our business, results of operations and prospects.
Our plan to achieve marketing approval of REM-001 Therapy depends partly on the accuracy of its preliminary efficacy analysis of REM-001 Therapy CMBC study data. While we believe the results of our preliminary efficacy analysis accurately reflect the actual clinical study results, a detailed analysis overseen by regulatory experts may yield different results.
We plan to utilize existing REM-001 Therapy clinical study data as supportive data when seeking marketing approval of REM-001 Therapy for the treatment of CMBC. Between February 1996 and January 1999, Miravant, with support from certain corporate partners, conducted four clinical studies for the treatment of CMBC using REM-001 Therapy. As part of our review of REM-001 Therapy’s data package, we noted that while Miravant’s investigators had done a safety analysis of all treated patients, these reports indicated an efficacy analysis was only performed on two of their four clinical studies. Notably, there had been no efficacy analysis on the other two studies which constituted approximately half of the CMBC patients who were treated with REM-001 Therapy. We originally performed a preliminary efficacy analysis on the data from all four CMBC studies, including the two that had not previously been analyzed. We then engaged regulatory experts who were either former FDA employees with directly related experience in reviewing similar oncology treatments who were then acting as independent consultants or individuals who have provided senior regulatory guidance to major pharmaceutical or medical device companies in situations that led to regulatory approval. These individuals guided us in conducting a second more in-depth analysis that yielded results consistent with our original analysis. Following that, we compiled a briefing document and submitted questions to FDA. While we believe the results of our preliminary efficacy analysis, and subsequent analysis conducted under the guidance of these experts which was consistent with its original preliminary analysis, accurately reflect the actual clinical study results and that the age of the underlying data from the clinical studies is not material, a more in-depth review may yield different conclusions. Such differing results may negatively impact our ability to pursue or achieve, or result in delays to obtain, marketing approval of REM-001 Therapy. There can be no certainty that results from our analyses done to date or results from future analyses that we may undertake will be sufficiently complete to satisfy FDA requests or that any results will be favorable to us.
We intend to use laser light devices that the FDA finds to be functionally equivalent to the Miravant devices in our planned clinical studies. If we are unable to demonstrate functional equivalence between the Miravant device and our intended laser light device or if the FDA refuses to allow the use of our intended laser light device, we will be unable to complete clinical studies for REM-001 Therapy and our business will be materially impaired.
Our REM-001 Therapy product consists of three parts, the DD series laser light source (or equivalent), the ML2-0400 light delivery device (or equivalent) and the drug REM-001. Pursuant to the Miravant oncology IND, the FDA previously approved all three components to be used together in certain Miravant CMBC Studies. Our plan is to use new lasers that are functionally equivalent to the Miravant DD2, the laser used in certain prior Miravant clinical studies, for CMBC. The light delivery devices we plan to use in our CMBC program are the same basic design developed and used previously by Miravant in its clinical studies. Our plan is to have clinical light delivery devices built by a contract medical device manufacturer using the basic Miravant design and tested to the same performance specifications as used previously. If the FDA finds that our intended laser light device is not functionally equivalent to the Miravant devices, the FDA may not approve any marketing application for REM-001.
Our REM-001 Therapy clinical study data may not be deemed acceptable by the FDA to support our new drug applications.
In seeking regulatory approval for REM-001, we intend to rely at least in part upon data gathered by Miravant in its initial Phase 1 studies and in four later Phase 2/3 clinical studies that were conducted approximately 20 years ago. Based on our initial interactions with the FDA, we believe the agency will accept these results as supportive data but we cannot ultimately be certain that the FDA will
46
accept data that old to support our new drug applications. Also based on our initial interactions with the FDA, we believe our plans for manufacturing investigational test materials will lead to investigational test materials that FDA will recognize as being sufficiently comparable to Miravant’s materials and also suitable for further investigational studies but FDA may later raise questions about the similarity of Miravant’s investigational testing material versus its manufactured investigational testing material, or may raise questions about the processes and methods under which this old data was collected or may raise additional concerns regarding the elapsed time period. If the FDA does not accept this data, we will have to incur significant costs which may require additional capital to redo some or all of the Miravant studies or supplement these studies with additional studies.
We may not receive regulatory approvals for our product candidates or there may be a delay in obtaining such approvals.
Our products and our ongoing development activities are subject to regulation by regulatory authorities in the countries in which we or our collaborators and distributors wish to test, manufacture or market our products. For instance, the FDA will regulate the product in the U.S. and equivalent authorities, such as the EMA, will regulate in Europe. Regulatory approval by these authorities will be subject to the evaluation of data relating to the quality, efficacy and safety of the product for its proposed use, and there can be no assurance that the regulatory authorities will find our data sufficient to support product approval of VAL-083 or any future product candidates.
The time required to obtain regulatory approval varies between countries. The FDA is required to facilitate the development and expedite the review of drugs and biologics that are intended for the treatment of a serious or life-threatening disease or condition and which demonstrate the potential to address unmet medical needs for the condition. Filling an unmet medical need is defined as providing a therapy where none exists or providing a therapy that may be potentially better than available therapy. Under the fast track program, the sponsor of a new drug or biologic candidate may request the FDA to designate the product for a specific indication as a fast track product concurrent with or after the filing of the IND for the product candidate. The FDA must determine if the product candidate qualifies for FTD within 60 days after receipt of the sponsor’s request. In the U.S., for products without “Fast Track” status, it can take over eighteen (18) months after submission of an application for product approval to receive the FDA’s decision. Even with FTD, FDA review and decision can take over twelve (12) months.
In December 2017, the FDA granted FTD for VAL-083 in patients with recurrent GBM and in June 2022, the FDA granted FTD for VAL-083 for the treatment of patients with newly-diagnosed unmethylated GBM.
Different regulators may impose their own requirements and may refuse to grant, or may require additional data before granting, an approval, notwithstanding that regulatory approval may have been granted by other regulators. Regulatory approval may be delayed, limited or denied for a number of reasons, including insufficient clinical data, the product not meeting safety or efficacy requirements or any relevant manufacturing processes or facilities not meeting applicable requirements as well as case load at the regulatory agency at the time.
We may fail to comply with regulatory requirements.
Our success will be dependent upon our ability, and our collaborative partners’ abilities, to maintain compliance with regulatory requirements, including cGMP, and safety reporting obligations. The failure to comply with applicable regulatory requirements can result in, among other things, fines, injunctions, civil penalties, total or partial suspension of regulatory approvals, refusal to approve pending applications, recalls or seizures of products, operating and production restrictions and criminal prosecutions.
Even if one of our product candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success and the market opportunity for the product candidate may be smaller than our estimates.
We have never commercialized a product. Even if VAL-083, REM-001, or any other product candidates is approved by the appropriate regulatory authorities for marketing and sale, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. For example, physicians are often reluctant to switch their patients from existing therapies even when new and potentially more effective or convenient treatments enter the market. Further, patients often acclimate to the therapy that they are currently taking and do not want to switch unless their physicians recommend switching products or they are required to switch therapies due to lack of reimbursement for existing therapies.
Efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may not be successful. If our product candidates are approved but do not achieve an adequate level of market
47
acceptance, we may not generate significant revenues and we may not become profitable. The degree of market acceptance of VAL-083, REM-001, or any other product candidate, if approved for commercial sale, will depend on a number of factors, including:
The potential market opportunities for our product candidates are difficult to estimate precisely. Our estimates of the potential market opportunities are predicated on many assumptions, including industry knowledge and publications, third-party research reports and other surveys. While we believe that our internal assumptions are reasonable, these assumptions involve the exercise of significant judgment on the part of our management, are inherently uncertain and the reasonableness of these assumptions has not been assessed by an independent source. If any of the assumptions proves to be inaccurate, the actual markets for our product candidate could be smaller than our estimates of the potential market opportunities.
If one of our product candidates receives marketing approval and we, or others, later discover that the drug is less effective than previously believed or causes undesirable side effects that were not previously identified, our ability to market the drug could be compromised.
Clinical studies of our product candidates are conducted in carefully defined subsets of patients who have agreed to enter into clinical studies. Consequently, it is possible that our clinical studies may indicate an apparent positive effect of a product candidate that is greater than the actual positive effect, if any, or alternatively fail to identify undesirable side effects. If, following approval of one of our product candidates, we, or others, discover that the drug is less effective than previously believed or causes undesirable side effects that were not previously identified, any of the following adverse events could occur:
48
Any of these events could have a material and adverse effect on our operations and business and could adversely impact our stock price.
Any product candidate for which we obtain marketing approval, along with the manufacturing processes, qualification testing, post-approval clinical data, labeling and promotional activities for such product, will be subject to continual and additional requirements of the FDA and other regulatory authorities.
These requirements include submissions of safety and other post-marketing information, reports, registration and listing requirements, good manufacturing practices, or GMP requirements relating to quality control, quality assurance and corresponding maintenance of records and documents, and recordkeeping. Even if marketing approval of any of our product candidates is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. The FDA closely regulates the post-approval marketing and promotion of pharmaceutical products to ensure such products are marketed only for the approved indications and in accordance with the provisions of the approved labeling.
In addition, later discovery of previously unknown problems with our products, manufacturing processes, or failure to comply with regulatory requirements, may lead to various adverse results, including:
If we are unable to establish sales, marketing and distribution capabilities or enter into acceptable sales, marketing and distribution arrangements with third parties, we may not be successful in commercializing any product candidates that we develop, if and when those product candidates are approved.
We do not have a sales, marketing or distribution infrastructure and have limited experience in the sale, marketing or distribution of pharmaceutical products. To achieve commercial success for any approved product, we must either develop a sales and marketing organization, outsource these functions to third parties, or license our product candidates to others. If approved, we may seek to license VAL-083 or REM-001 to a large pharmaceutical company with greater resources and experience than us. We may not be able to license VAL-083 or REM-001 on reasonable terms, if at all. The development of sales, marketing and distribution capabilities will require substantial resources, will be time-consuming and could delay any product launch. We expect that we will commence the development of these capabilities prior to receiving approval of our product candidates. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing and distribution capabilities is delayed or does not occur for any reason, we could have prematurely or unnecessarily incurred these commercialization costs. Such a delay may be costly, and our investment could be lost if we cannot retain or reposition our sales and marketing personnel. In addition, we may not be able to hire or retain a sales force in the United States that is sufficient in size or has adequate expertise in the medical markets that we plan to target. If we are unable to establish or retain a sales force and marketing and distribution capabilities, our operating results may be adversely affected. If a potential partner has development or commercialization expertise that we believe is particularly relevant to our product candidates, then we may seek to collaborate with that potential partner even if we believe we could otherwise develop and commercialize the product independently.
49
We expect to seek one or more strategic partners for commercialization of our product candidates outside the United States. As a result of entering into arrangements with third parties to perform sales, marketing and distribution services, our product revenues or the profitability of these product revenues may be lower, perhaps substantially lower, than if we were to directly market and sell products in those markets. Furthermore, we may be unsuccessful in entering into the necessary arrangements with third parties or may be unable to do so on terms that are favorable to us. In addition, we may have little or no control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively.
If we do not establish sales and marketing capabilities, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates.
We face substantial competition from other pharmaceutical and biotechnology companies and our operating results may suffer if we fail to compete effectively.
The development and commercialization of new drug products is highly competitive. We expect that we will face significant competition from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide with respect to VAL-083, REM-001, and any other product candidates that we may seek to develop or commercialize in the future. Specifically, due to the large unmet medical need, global demographics and relatively attractive reimbursement dynamics, the oncology market is fiercely competitive and there are a number of large pharmaceutical and biotechnology companies that currently market and sell products or are pursuing the development of product candidates for the treatment of cancer. Our competitors may succeed in developing, acquiring or licensing technologies and drug products that are more effective, have fewer or more tolerable side effects or are less costly than any product candidates that we are currently developing or that we may develop, which could render our product candidates obsolete and noncompetitive. All of the top ten global pharmaceutical companies and many of the mid-size pharmaceutical companies have a strong research and development and commercial presence in oncology. Several companies are marketing and developing oncology immunotherapy products.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other marketing approval for their products before we are able to obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
Many of our existing and potential future competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical studies, obtaining marketing approvals and marketing approved products than our does. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical study sites and patient registration for clinical studies, as well as in acquiring technologies complementary to, or necessary for, our programs.
If we are unable to obtain, or are delayed in obtaining, state regulatory licenses for the distribution of our products, we would not be able to sell our product candidates.
The majority of states require manufacturer and/or wholesaler licenses for the sale and distribution of drugs into that state. The application process is complicated, time consuming and requires dedicated personnel or a third-party to oversee and manage. If we are delayed in obtaining these state licenses, or denied the licenses, even with FDA approval, we would not be able to sell or ship product into that state which would adversely affect our sales and revenues.
We rely on key personnel and members of management and, if we are unable to retain or motivate key personnel or management, or hire qualified personnel, we may not be able to grow effectively.
We are dependent on certain members of our management, scientific and drug development staff and consultants, the loss of services of one or more of whom could materially adversely affect us.
We currently have three full-time employees, and retain the services of approximately 20 persons on an independent contractor/consultant and contract-employment basis. Our ability to manage growth effectively will require us to continue to implement and improve our management systems and to recruit and train new employees. Although we have done so in the past and expect to do so in the future, there can be no assurance that we will be able to successfully attract and retain skilled and experienced personnel.
50
Our success depends in large part upon our ability to attract and retain highly qualified personnel. We compete in our hiring efforts with other pharmaceutical and biotechnology companies, as well as universities and nonprofit research organizations, and we may have to pay higher salaries to attract and retain personnel, which would be very costly.
We may be subject to foreign exchange fluctuation.
Our functional and reporting currency is the United States dollar. We maintain bank accounts in United States and Canadian dollars. A portion of our expenditures are in foreign currencies, most notably in Canadian dollars and Euros, and therefore we are subject to foreign currency fluctuations, which may, from time to time, impact our financial position and results. We may enter into hedging arrangements under specific circumstances, typically through the use of forward or futures currency contracts, to minimize the impact of increases in the value of the Canadian dollar. In order to minimize our exposure to foreign exchange fluctuations we may hold sufficient Canadian dollars to cover our expected Canadian dollar expenditures.
Product liability lawsuits against us could divert our resources, cause us to incur substantial liabilities and limit commercialization of any products that we may develop.
We face an inherent risk of product liability claims as a result of the clinical testing of our product candidates despite obtaining appropriate informed consents from our clinical study participants. We will face an even greater risk if we commercially sell any product that we may develop. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Regardless of the merits or eventual outcome, liability claims may result in:
Although we maintain general liability insurance, this insurance may not fully cover potential liabilities that we may incur. The cost of any product liability litigation or other proceeding, even if resolved in our favor, could be substantial. We will need to increase our insurance coverage if and when we begin selling any product candidate that receives marketing approval. In addition, insurance coverage is becoming increasingly expensive. If we are unable to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product liability claims, it could prevent or inhibit the development and commercial production and sale of our product candidates, which could adversely affect our business, financial condition, results of operations and prospects.
Risks Related to Our Dependence on Third Parties
We rely on third parties to conduct clinical studies for our product candidate. Any failure by a third-party to meet our obligations with respect to the clinical development of our product candidate may delay or impair our ability to obtain regulatory approval for our product candidate.
We rely on academic institutions and private oncology centers to conduct our clinical studies. Our reliance on third parties to conduct clinical studies could, depending on the actions of such third parties, jeopardize the validity of the clinical data generated and adversely affect our ability to obtain marketing approval from the FDA or other applicable regulatory authorities.
Such clinical study arrangements provide us with information rights with respect to the clinical data, including access to and the ability to use and reference the data, including for our own regulatory filings, resulting from the clinical studies. If investigators or institutions breach their obligations with respect to the clinical studies of our product candidate, or if the data proves to be inadequate, then our ability to design and conduct any future clinical studies may be adversely affected.
51
We rely, and expect to continue to rely, on third parties to conduct our clinical studies, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such studies.
We currently rely on third-party clinical research organizations, or CROs, to conduct our clinical studies. We expect to continue to rely on third parties, such as CROs, clinical data management organizations, medical institutions and clinical investigators, to conduct our clinical studies. Our agreements with these third parties generally allow the third-party to terminate the agreement at any time. If we are required to enter into alternative arrangements because of any such termination the introduction of our product candidates to market could be delayed.
Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, we design our clinical studies and will remain responsible for ensuring that each of our clinical studies are conducted in accordance with the general investigational plan and protocols for the study. Moreover, the FDA requires us to comply with standards, commonly referred to as good clinical practices, or GCPs, for conducting, recording and reporting the results of clinical studies to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of study participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. We also are required to register ongoing clinical studies and post the results of completed clinical studies on a government-sponsored database, Clinicaltrials.gov, within specified timeframes. Failure to do so can result in fines, adverse publicity and civil and criminal sanctions.
Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our clinical studies in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for our product candidates and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates.
We also expect to rely on other third parties to store and distribute drug supplies for our clinical studies. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidate or commercialization of our products, producing additional losses and depriving us of potential product revenue.
We may seek to enter into collaborations with third parties for the development and commercialization of our product candidates. If we fail to enter into such collaborations, or such collaborations are not successful, we may not be able to capitalize on the market potential of our product candidates.
We may seek third-party collaborators for development and commercialization of our product candidates. Our likely collaborators for any marketing, distribution, development, licensing or broader collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies, non-profit organizations, government agencies, and biotechnology companies. We are currently party to a limited number of such arrangements and have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidate. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations involving our product candidates currently pose, and will continue to pose, the following risks to us:
52
Collaboration agreements may not lead to development or commercialization of our product candidates in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
If we are not able to establish collaborations, we may have to alter our development and commercialization plans.
Our drug development programs and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. We may decide to collaborate with pharmaceutical and biotechnology companies for the development and potential commercialization of our product candidates.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of preclinical studies or clinical studies, the likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidate. We may also be restricted under future license agreements from entering into agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of our product candidates, reduce or delay our development program, delay our potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our product candidate or bring it to market and generate product revenue.
We currently manufacture VAL-083 at a single location. Any disruption at this facility could adversely affect our business and results of operations.
We have engaged a single manufacturer to produce VAL-083 GMP active pharmaceutical ingredient and a single manufacturer to produce VAL-083 drug product for our clinical studies. In addition, we previously have relied on our manufacturing partner, Guangxi Wuzhou Pharmaceutical Company, for the manufacture of clinical supply of VAL-083 for our preclinical studies as well as our now-completed Phase 2 clinical study conducted in China. If our manufacturer’s facilities were damaged or destroyed, or otherwise subject to disruption, it would require substantial lead-time to replace our clinical supply. In such event, we would be forced to rely entirely on other third-party contract manufacturers for an indefinite period of time. We do not currently have established relationships with any back-up manufacturers. At this time no drug product has been manufactured by a third-party back-up manufacturer. Any disruptions or delays by our third-party manufacturers or Guangxi Wuzhou Pharmaceutical Company or their failure to meet regulatory compliance could impair our ability to develop VAL-083, which would adversely affect our business and results of operations.
53
We rely on these third-party manufacturers to provide drug product supply for all of our clinical studies for VAL-083. There is no assurance that such a supplier will be able to meet our needs from a technical, timing, or cost-effective manner. Our failure to execute appropriate agreements with such a third-party manufacturer would delay, or halt, our clinical studies.
We do not have a clinical supply of REM-001 and we do not have our own manufacturing facilities. If a third-party manufacturer fails to meet applicable regulatory requirements or to supply us for any reason, we will be unable to complete clinical studies for REM-001 Therapy and our business will be materially impaired.
We have engaged manufacturers to produce REM-001 GMP pharmaceutical ingredient for our planned clinical studies. Our plan calls for the use third-party manufacturers to produce the product for us. If and when approved, we intend to have a third-party manufacture commercial supplies of the product as well. We have not yet completed the transfer of the technology or manufactured the product at these facilities and our failure to timely do so will delay the commencement of our clinical studies and may also impact the timing for the submission of our NDA for REM-001 Therapy.
We rely on these third-party manufacturers to provide drug product supply for all of our planned clinical studies for REM-001. There is no assurance that such suppliers will be able to meet our needs from a technical, timing, or cost-effective manner. Our failure to execute appropriate agreements with such a third-party manufacturer would delay, or halt, our clinical studies.
We do not have a clinical supply of light delivery devices for use with REM-001 Therapy. Moreover, we do not have our own manufacturing facilities nor have we contracted a third-party to manufacture these devices for us. If a third-party manufacturer fails to meet applicable regulatory requirements or to supply us for any reason, we will be unable to complete clinical studies for REM-001 Therapy and our business will be materially impaired.
We do not have a clinical supply of REM-001 Therapy light delivery devices. Our plan calls for the use of a third-party manufacturer to produce these devices for us. The failure of a third-party manufacturer to supply such devices in a timely and cost-effective manner will delay the commencement of our clinical studies and may also impact the timing for the submission of our NDA for REM-001 Therapy.
We are planning to use laser light devices that the FDA finds to be functionally equivalent to the Miravant devices in our planned clinical studies. We do not have our own manufacturing facilities for conducting these activities nor have we contracted a third-party to manufacture these devices for us. If we are unable to contract a third-party manufacturer, or if a third-party manufacturer fails to meet applicable regulatory requirements or to supply it for any reason, we will be unable to complete clinical studies for REM-001 Therapy and our business will be materially impaired.
Our plan relies on using laser light devices that the FDA finds to be functionally equivalent to the Miravant devices. Our plan calls for the use of a third-party manufacturer to produce new laser devices for us. The failure of a third-party manufacturer to supply such devices in a timely and cost-effective manner will delay the commencement of our clinical studies and may also impact the timing for the submission of our NDA for REM-001 Therapy.
We may become subject to liabilities related to risks inherent in working with hazardous materials.
Our discovery and development processes involve the controlled use of hazardous and radioactive materials. We are subject to federal, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by such laws and regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result and any such liability could exceed our resources. We are not specifically insured with respect to this liability. Although we believe that we are in compliance in all material respects with applicable environmental laws and regulations and currently do not expect to make material capital expenditures for environmental control facilities in the near-term, there can be no assurance that we will not be required to incur significant costs to comply with environmental laws and regulations in the future, or that our operations, business or assets will not be materially adversely affected by current or future environmental laws or regulations.
Risks Related to Our Common Stock
The market price of our common stock is, and is likely to continue to be, highly volatile and subject to wide fluctuations.
The market price of our common stock is highly volatile and could be subject to wide fluctuations in response to a number of factors that are beyond our control, including:
54
Our Articles allow for our board of directors to create new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our common stock.
Our board of directors has the authority to fix and determine the relative rights and preferences of preferred stock. Our board of directors has the authority to issue up to 5,000,000 shares of our preferred stock (of which 278,530 shares have been designated Series A Preferred Stock and are issued and outstanding as of June 30, 2022 and 1,000,000 shares have been designated as Series B Preferred Stock of which none are issued and outstanding as of June 30, 2022) without further stockholder approval. In addition, 28,400 have been designated as Series C (22,000 as Series C-1, 2,700 as Series C-2, and 3,700 as Series C-3) of which 16,838 are issued and outstanding as of June 30, 2022. As a result, our board of directors could authorize the issuance of additional series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of common stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our common stock. In addition, our board of directors could authorize the issuance of a series of preferred stock that has greater voting power than our common stock, or that is convertible into our common stock, which could decrease the relative voting power of our common stock or result in dilution to our existing stockholders. Although we have no present intention to issue any additional shares of preferred stock or to create any additional series of preferred stock, we may issue such shares in the future.
Issuance of our common stock upon exercise of warrants or options, or conversion of the Series C Preferred Stock may depress the price of our common stock.
As of September 26, 2022, we had 80,807 shares of common stock issued and outstanding, outstanding warrants to purchase 35,704 shares of common stock, warrants to purchase 2.4 shares of our Series C Preferred Stock that upon exercise are convertible into 2,100 shares of common stock, outstanding stock options to purchase 12,291 shares of common stock, 17 outstanding shares of Series C Preferred Stock that are convertible into 14,497 shares of common stock. All common stock warrants and stock options are convertible, or exercisable into, one share of common stock. The Series C Preferred Stock (issued in three series) is convertible into shares of common stock at $1.16 per share (Series C-1), $1.214 per share (Series C-2) or $1.15 per share (Series C-3), respectively. The Series C Preferred stock purchase warrants are convertible into Series C Preferred Stock at $1,000 per share for either Series C-1, Series C-2, or Series C-3 Preferred Stock, as applicable.
The issuance of shares of our common stock upon the exercise of outstanding warrants or options, or the conversion of our Series C-1, C-2, and C-3 Series Preferred Stock, could result in substantial dilution to our stockholders, which may have a negative effect on the price of our common stock.
We do not intend to pay cash dividends on our common stock for the foreseeable future.
We have paid no cash dividends on our common stock to date and we do not anticipate paying any dividends to holders of our common stock in the foreseeable future. While our future dividend policy will be based on the operating results and capital needs of the business, we currently anticipate that any earnings will be retained to finance our future expansion and for the implementation of our business plan. Investors should take note of the fact that a lack of a dividend can further affect the market value of our common stock, and could significantly affect the value of any investment in us.
55
FORWARD-LOOKING STATEMENTS
This annual report on Form 10-K contains forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 under Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements include statements with respect to our beliefs, plans, objectives, goals, expectations, anticipations, assumptions, estimates, intentions and future performance, and involve known and unknown risks, uncertainties and other factors, which may be beyond our control, and which may cause our actual results, performance or achievements to be materially different from future results, performance or achievements expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be forward-looking statements. You can identify these forward-looking statements through our use of words such as “may,” “can,” “anticipate,” “assume,” “should,” “indicate,” “would,” “believe,” “contemplate,” “expect,” “seek,” “estimate,” “continue,” “plan,” “point to,” “project,” “predict,” “could,” “intend,” “target,” “potential” and other similar words and expressions of the future.
There are a number of important factors that could cause the actual results to differ materially from those expressed in any forward-looking statement made by us. These factors include, but are not limited to:
The foregoing does not represent an exhaustive list of matters that may be covered by the forward-looking statements contained herein or risk factors that we are faced with that may cause our actual results to differ from those anticipated in our forward-looking statements. Please see “Risk Factors” in this Annual Report on Form 10-K under Part I, Item 1A, for additional risks which could adversely impact our business and financial performance.
All forward-looking statements are expressly qualified in their entirety by this cautionary notice. You are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this report or the date of the document incorporated by reference into this report. We have no obligation, and expressly disclaim any obligation, to update, revise or correct any of the forward-looking statements, whether as a result of new information, future events or otherwise. We have expressed our expectations, beliefs and projections in good faith and we believe they have a reasonable basis. However, we cannot assure you that our expectations, beliefs or projections will result or be achieved or accomplished.
56
Item 1B. Unresolved Staff Comments.
Not required for a smaller reporting company.
Item 2. Properties.
Our corporate headquarters are currently located at 9920 Pacific Heights Blvd, Suite 150, San Diego CA, 92121. The current rent at that location under a one-year renewable lease is $2.4 thousand per year. We also rent our administrative offices located at Suite 720-999 West Broadway, Vancouver, British Columbia, Canada on a month-to-month basis at a rate of $3.4 thousand (CA$4.4 thousand) per month. During the year ended June 30, 2022, we recorded a total of $41.4 thousand as rent expense (2021 - $40 thousand).
In addition, Valent, which is owned by Dr. Dennis Brown, our Chief Scientific Officer, leases facilities in California and we have access to such facilities pursuant to an informal unwritten arrangement with Valent. Our leased premises, academic relationships, and access to the Valent facility are sufficient to meet the immediate needs of our business, research and operations.
Item 3. Legal Proceedings.
There are no legal proceedings to which we are a party or any of our property is the subject.
Item 4. Mine Safety Disclosures.
Not applicable.
57
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Since August 20, 2020, our common stock has been listed on The Nasdaq Capital Market LLC under the symbol “KTRA”. From July 12, 2016, until August 19, 2020, our common stock was listed on The Nasdaq Capital Market LLC under the symbol “DMPI”. Previously, our common stock was quoted on the OTC.QX, and prior to that, on the OTCQB.
As of September 26, 2022, there were approximately 801 holders of record of our common stock.
Dividends
We have never declared or paid any cash dividends on our common stock. We currently intend to retain future earnings, if any, to finance the expansion of our business. As a result, we do not anticipate paying any cash dividends in the foreseeable future.
Sales of Unregistered Securities
None.
Issuer Purchases of Equity Securities
None.
Item 6. Reserved
58
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
This Management’s Discussion and Analysis (“MD&A”) contains “forward-looking statements”, within the meaning of the Private Securities Litigation Reform Act of 1995, which represent our projections, estimates, expectations, or beliefs concerning, among other things, financial items that relate to management’s future plans or objectives or to our future economic and financial performance. In some cases, you can identify these statements by terminology such as “may”, “should”, “plans”, “believe”, “will”, “anticipate”, “estimate”, “expect” “project”, or “intend”, including their opposites or similar phrases or expressions. You should be aware that these statements are projections or estimates as to future events and are subject to a number of factors that may tend to influence the accuracy of the statements. These forward-looking statements should not be regarded as a representation by us or any other person that our events or plans will be achieved. You should not unduly rely on these forward-looking statements, which speak only as of the date of this report. Except as may be required under applicable securities laws, we undertake no obligation to publicly revise any forward-looking statement to reflect circumstances or events after the date of this report or to reflect the occurrence of unanticipated events.
You should review the factors and risks we describe under “Risk Factors” in this report on Form 10-K for the year ended June 30, 2022 and in our other filings with the Securities and Exchange Commission, available at www.sec.gov. Actual results may differ materially from any forward-looking statement.
Impact of Coronavirus (“COVID-19”) on our Operations, Financial Condition, Liquidity and Results of Operations
In December 2019 a novel strain of coronavirus, COVID-19 was reported to have surfaced in Wuhan, China and on March 11, 2020, it was declared a pandemic by the World Health Organization. The ultimate impact of the COVID-19 pandemic on our operations is unknown and will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration of the COVID-19 outbreak, new information which may emerge concerning the duration and severity of the COVID-19 pandemic, and any additional preventative and protective actions that governments, or us, may determine are needed.
The COVID-19 pandemic did not cause significant disruption to our Phase 2 clinical studies. Each of our now-completed Phase 2 clinical studies was conducted at respective single sites which reduced the risk of study disruption. Any disruptions to patient treatments for our Phase 2 studies were within allowances under each study protocol. Access to the sites by our clinical monitors was limited during the COVID-19 pandemic but the recording of study data in both studies and patient treatments at both study sites was conducted per protocol.
Regarding the VAL-083 study arm of the GBM AGILE Study that is currently being conducted at multiple sites in the United States, Canada and Europe, we have not experienced any significant impacts on patient enrollment or treatment. With respect to the REM-001 drug supply, we are currently experiencing some delays in contract manufacturing schedules and supplies which we attribute to COVID-19. The current delays could have an impact on our REM-001 program timeline.
Including aggregate net proceeds of approximately $21.6 million received from two registered direct financings that closed on September 28, 2021 and April 14, 2022, respectively, as well as the potential proceeds available under our Purchase Agreement with Lincoln Park, we estimate that we have cash available to fund planned operations for less than one year from the date of issuance of our June 30, 2022 consolidated financial statements. The COVID-19 pandemic has created significant economic uncertainty and volatility in the credit and capital markets. The ultimate impact of the COVID-19 pandemic on our ability to raise additional capital is unknown and will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration of the COVID-19 outbreak and new information which may emerge concerning the severity of the COVID-19 pandemic. We may not be able to raise sufficient additional capital and may tailor our drug candidate development programs based on the amount of funding we are able to raise in the future. Nevertheless, there is no assurance that these initiatives will be successful.
Corporate History
We are a Nevada corporation formed on June 24, 2009, under the name Berry Only, Inc. On January 25, 2013, we entered into and closed the Exchange Agreement with Del Mar (BC), Callco, Exchangeco and the security holders of Del Mar (BC). Upon completion of the Exchange Agreement, Del Mar (BC) became our wholly-owned subsidiary which we refer to as the Reverse Acquisition.
On June 10, 2020, we entered into the Merger Agreement by and among Merger Sub and Adgero. On August 19, 2020, upon the terms, and subject to the conditions set forth in the Merger Agreement, Merger Sub merged with and into Adgero, the separate corporate existence of Merger Sub ceased, and Adgero continued its existence under Delaware law as the surviving corporation in the
59
Merger and became our direct, wholly-owned subsidiary. As a result of the Merger, each issued and outstanding share of Adgero common stock (other than treasury shares held by Adgero), was converted automatically into the right to receive 1.5740 shares, or the Exchange Ratio, of our common stock, and cash in lieu of any fractional shares. Also, each outstanding warrant to purchase Adgero Common Stock was converted into a warrant exercisable for that number of shares of our common stock equal to the product of (x) the aggregate number of shares of Adgero Common Stock for which such warrant was exercisable and (y) the Exchange Ratio.
Following the completion of the Merger, we changed our name from DelMar Pharmaceuticals, Inc. to Kintara Therapeutics, Inc. In conjunction with the closing of the Merger, and through a series of three private placement closings, we issued a total of 25,028 shares of Series C Preferred Stock at a purchase price of $1,000 per share for total aggregate gross proceeds of $25 million, or net proceeds of approximately $21.6 million.
The Series C Stock was issued in three series at conversion prices equal to $1.16, $1.214 and $1.15, respectively. As a result, we issued a total of 25,028 shares of Series C Stock, which are convertible into an aggregate 21,516,484 shares of common stock. As part of the private placement, we issued Placement Agent Warrants to purchase 2,504 shares of Series C Stock to the placement agent. The Placement Agent Warrants have an exercise price of $1,000 per share, provide for a cashless exercise feature and are exercisable for a period of four years from the date of the initial closing of the private placement. The Series C Stock and the shares of Series C Stock issuable upon exercise of the Placement Agent Warrants will be entitled to receive dividends, payable in shares of our common stock, at a rate of 10%, 15%, 20%, and 25%, of the number of shares of common stock issuable upon conversion of the Series C Stock, on the 12th, 24th, 36th and 48th month anniversary of the initial closing of the private placement, which occurred on August 19, 2020, provided that the holder of such shares has not converted the shares of Series C Stock prior to the applicable dividend rate.
Outstanding Securities
As of September 26, 2022, we had 80,807 shares of common stock issued and outstanding, outstanding warrants to purchase 35,704 shares of common stock, warrants to purchase 2.4 shares of our Series C Preferred Stock that upon exercise are convertible into 2,100 shares of common stock, outstanding stock options to purchase 12,291 shares of common stock, 17 outstanding shares of Series C Preferred Stock that are convertible into 14,497 shares of common stock. All common stock warrants and stock options are convertible, or exercisable into, one share of common stock. The Series C Preferred Stock (issued in three series) is convertible into shares of common stock at $1.16 per share (Series C-1), $1.214 per share (Series C-2) or $1.15 per share (Series C-3), respectively. The Series C Preferred stock purchase warrants are convertible into Series C Preferred Stock at $1,000 per share for either Series C-1, Series C-2, or Series C-3 Preferred Stock, as applicable.
On June 21, 2022, we amended our articles of incorporation, as amended, to increase the number of authorized shares of common stock from 175,000,000 to 275,000,000 shares.
Related Parties
We acquired our initial patents and technology rights from Valent, an entity owned by Dr. Dennis Brown, our Chief Scientific Officer. As a result, Valent is a related party.
Selected Annual Information
The financial information reported herein has been prepared in accordance with accounting principles generally accepted in the United States. Our functional currency at June 30, 2022 and June 30, 2021 is the US$ and our financial data is expressed in thousands, except par value and per share amounts unless otherwise noted. The following tables represent selected financial information for us for the periods presented.
Selected Balance Sheet data (in thousands)
|
|
June 30, |
|
|
June 30, |
|
||
|
|
$ |
|
|
$ |
|
||
Cash and cash equivalents |
|
|
11,780 |
|
|
|
10,537 |
|
Working capital |
|
|
9,268 |
|
|
|
9,013 |
|
Total assets |
|
|
15,948 |
|
|
|
13,543 |
|
Total stockholders’ equity |
|
|
11,795 |
|
|
|
10,581 |
|
60
Selected Statement of Operations data (in thousands, except per share data)
For the years ended
|
|
June 30, |
|
|
June 30, |
|
||
Expenses |
|
|
|
|
|
|
||
Research and development |
$ |
|
15,173 |
|
$ |
|
11,815 |
|
General and administrative |
|
|
7,509 |
|
|
|
9,757 |
|
Merger costs |
|
|
— |
|
|
|
500 |
|
In-process research and development |
|
|
— |
|
|
|
16,094 |
|
|
|
|
(22,682 |
) |
|
|
(38,166 |
) |
Other income (loss) |
|
|
|
|
|
|
||
Foreign exchange |
|
|
7 |
|
|
|
(12 |
) |
Amortization of deferred loan costs |
|
|
— |
|
|
|
(94 |
) |
Interest - net |
|
|
14 |
|
|
|
(26 |
) |
|
|
|
21 |
|
|
|
(132 |
) |
Net loss for the year |
|
|
(22,661 |
) |
|
|
(38,298 |
) |
Deemed dividend recognized on beneficial conversion |
|
|
— |
|
|
|
(3,181 |
) |
Series A Preferred cash dividend |
|
|
(8 |
) |
|
|
(8 |
) |
Series B Preferred stock dividend |
|
|
— |
|
|
|
(17 |
) |
Series C Preferred stock dividend |
|
|
(2,462 |
) |
|
|
— |
|
Net loss for the year attributable to common |
$ |
|
(25,131 |
) |
$ |
|
(41,504 |
) |
Basic and fully diluted weighted average number of shares |
|
|
48,702 |
|
|
|
25,886 |
|
Basic and fully diluted loss per share |
$ |
|
(0.52 |
) |
$ |
|
(1.60 |
) |
Expenses net of non-cash, share-based compensation expense - non-GAAP
The following table discloses research and development, and general and administrative expenses net of non-cash, share-based compensation payment expense. The disclosure has been provided to reconcile the total operational expenses on a U.S. GAAP basis and the non-GAAP operational expenses net of non-cash, share-based compensation in order to provide an estimate of cash used in research and development, and general and administrative expense. Management uses this estimate of expenses net of non-cash, share-based compensation for forecasting and budget purposes to determine the allocation of resources and to plan for future financing opportunities.
For the years ended (in thousands)
|
|
June 30, |
|
|
June 30, |
|
||
Research and development - GAAP |
|
|
15,173 |
|
|
|
11,815 |
|
Less: non-cash share-based compensation expense |
|
|
(601 |
) |
|
|
(1,478 |
) |
Research and development net of non-cash share-based, |
|
|
14,572 |
|
|
|
10,337 |
|
|
|
|
|
|
|
|
||
General and administrative - GAAP |
|
|
7,509 |
|
|
|
9,757 |
|
Less: non-cash share-based compensation expense |
|
|
(1,682 |
) |
|
|
(4,367 |
) |
General and administrative net of non-cash share-based, |
|
|
5,827 |
|
|
|
5,390 |
|
61
Comparison of the years ended June 30, 2022 and June 30, 2021
|
|
Years ended |
|
|
|
|
|
|
|
|||||||
|
|
June 30, |
|
|
June 30, |
|
|
Change $ |
|
|
Change % |
|
||||
|
|
(in thousands) |
|
|
|
|
||||||||||
Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Research and development |
|
|
15,173 |
|
|
|
11,815 |
|
|
|
3,358 |
|
|
|
28 |
|
General and administrative |
|
|
7,509 |
|
|
|
9,757 |
|
|
|
(2,248 |
) |
|
|
(23 |
) |
Merger costs |
|
|
— |
|
|
|
500 |
|
|
|
(500 |
) |
|
|
(100 |
) |
In-process research and development |
|
|
— |
|
|
|
16,094 |
|
|
|
(16,094 |
) |
|
|
(100 |
) |
|
|
|
(22,682 |
) |
|
|
(38,166 |
) |
|
|
15,484 |
|
|
|
|
|
Other income (loss) |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Foreign exchange |
|
|
7 |
|
|
|
(12 |
) |
|
|
19 |
|
|
|
(158 |
) |
Amortization of deferred loan costs |
|
|
— |
|
|
|
(94 |
) |
|
|
94 |
|
|
|
(100 |
) |
Interest, net |
|
|
14 |
|
|
|
(26 |
) |
|
|
40 |
|
|
|
(154 |
) |
|
|
|
21 |
|
|
|
(132 |
) |
|
|
153 |
|
|
|
|
|
Net loss |
|
|
(22,661 |
) |
|
|
(38,298 |
) |
|
|
15,637 |
|
|
|
|
|
Research and Development
Research and development expenses increased to $15,173 for the year ended June 30, 2022, from $11,815 for the year ended June 30, 2021. The increase was largely attributable to higher clinical development costs partially offset by lower non-cash, share-based compensation incurred during the year ended June 30, 2022, compared to the year ended June 30, 2021, due primarily to the recognition of compensation expense for stock options granted in September 2020.
Clinical development costs have increased in the current year compared to the prior year largely due to expenses related to the GCAR GBM AGILE Study. Patient recruitment for this study commenced in January 2021 and ongoing study costs, including clinical site activation and patient enrollment, were incurred during the full year ended June 30, 2022 but were incurred for only a portion of the year ended June 30, 2021. In addition, during the current year we incurred compound manufacturing costs and initial study preparation costs relating to our REM-001, 15-patient clinical study which is planned to commence patient enrollment around the end of the third quarter of calendar 2022.
General and Administrative
General and administrative expenses were $7,509 for the year ended June 30, 2022, compared to $9,757 for the year ended June 30, 2021. We incurred lower non-cash, share-based compensation expenses in the current year compared to the prior year due to the recognition of compensation expense in the year ended June 30, 2021, for stock options granted in September 2020. In addition, professional fees were lower in the year ended June 30, 2022, compared to the year ended June 30, 2021.
Partially offsetting the lower non-cash, share-based compensation expenses and professional fees for the year ended June 30, 2022, were higher personnel, and office and sundry costs. Personnel costs increased due to the recognition of employee severance costs during the current year while office and sundry has increased due to higher insurance costs.
Merger Costs
We incurred costs related to the Adgero Merger of $500 for the year ended June 30, 2021, and none in the year ended June 30, 2022. All of these costs have been expensed.
Acquired In-Process Research and Development Expense
We acquired in-process research and development assets in connection with the Adgero Merger. As the acquired in-process research and development assets were deemed to have no current or alternative future use, an expense of $16,094 was recognized in the consolidated statements of operations for the year ended June 30, 2021.
Preferred Stock Dividends
During the year ended June 30, 2022, we issued 1,698 (2021 – nil) shares of common stock as a stock dividend on the Series C Preferred stock and recognized $2,462 (2021 - nil) as a direct increase in accumulated deficit.
For each of the years ended June 30, 2022, and 2021, we recorded $8 related to the dividend payable to Valent on the Series A Preferred Stock. The dividend has been recorded as a direct increase in accumulated deficit for both years.
62
During the year ended June 30, 2022, we issued nil (2021 – 11) shares of common stock as a dividend on the Series B Preferred stock and recognized nil (2021 - $17) as a direct increase in accumulated deficit.
Liquidity and Capital Resources
Comparison of the years ended June 30, 2022 and June 30, 2021
|
|
June 30, |
|
|
June 30, |
|
|
Change |
|
|
Change |
|
||||
Cash flows from operating activities |
|
|
(20,392 |
) |
|
|
(18,860 |
) |
|
|
(1,532 |
) |
|
|
8 |
|
Cash flows from investing activities |
|
|
— |
|
|
|
964 |
|
|
|
(964 |
) |
|
|
(100 |
) |
Cash flows from financing activities |
|
|
21,635 |
|
|
|
26,041 |
|
|
|
(4,406 |
) |
|
|
(17 |
) |
Operating Activities
Net cash used in operating activities increased to $20,392 for the year ended June 30, 2022, from $18,860 for the year ended June 30, 2021. During the year ended June 30, 2022, and 2021, we reported net losses of $22,661 and $38,298, respectively. While the loss in the prior year was larger than the current year, the prior year included a non-cash amount of $16,094 relating to the recognition of acquired in-process research and development expense related to the Adgero Merger. Additional changes in adjustments to reconcile net loss to net cash used in operating activities for the year ended June 30, 2022, included stock option expense of $2,248 being recognized during the current year compared to $5,276 in the prior year. The most significant changes in working capital for the year ended June 30, 2022, were related to an increase in accounts payable and accrued liabilities of $1,007 and from an increase in prepaid expenses, deposits and other of $722. The most significant change in working capital for the year ended June 30, 2021, was from a use of cash due to an increase in prepaid expenses and deposits related primarily to a $2,600 payment to GCAR for study initiation and patient recruitment.
Investing Activities
There were no investing activities during the year ended June 30, 2022. During the year ended June 30, 2021, we acquired $969 in cash as part of the Adgero Merger that closed on August 19, 2020.
Financing Activities
During the year ended June 30, 2022, we received $21,569 in aggregate net proceeds from the completion of two registered direct financings that closed on September 28, 2021, and April 14, 2022, respectively. We also received $74 from the cash exercise of stock purchase warrants.
During the year ended June 30, 2021, we received $21,598 in net proceeds from the completion of a private placement of Series C Preferred stock. We also received $4,404 and $77 from the respective cash exercise of common stock purchase warrants and stock options. In addition, during the year ended June 30, 2021, we received proceeds from the NBTS Loan of $500 which we repaid during the prior year.
Going Concern and Capital Expenditure Requirements
Going Concern and Management Plans
(See note 1 to the consolidated financial statements)
The consolidated financial statements have been prepared on a going concern basis, which assumes that we will continue our operations for the foreseeable future and contemplates the realization of assets and the settlement of liabilities in the normal course of business.
For the year ended June 30, 2022, we reported a loss of $22,661 and a negative cash flow from operations of $20,392. We had an accumulated deficit of $136,356 and had cash and cash equivalents of $11,780 as of June 30, 2022. We are in the clinical stage and have not generated any revenues to-date. We do not have the prospect of achieving revenues until such time that our product candidates are commercialized, or partnered, which may not ever occur. On September 28, 2021, and April 14, 2022, respectively, we completed registered direct financings for aggregate net proceeds of approximately $21.6 million and on August 2, 2022, we entered into the Purchase Agreement with Lincoln Park, which subject to certain limitations, may allow us to sell shares our common stock for a total of up to $20 million in gross proceeds. Subsequent to June 30, 2022, we have sold 11,470 shares of common stock for total gross proceeds of approximately $1,976 under this facility. However, potential proceeds under the Purchase Agreement are uncertain as to timing and amount. Even with the proceeds from the two registered direct financings and the existence of the Purchase
63
Agreement, we will require additional funding to maintain our clinical trials, research and development projects, and for general operations. These circumstances indicate substantial doubt exists about our ability to continue as a going concern within one year from the date of filing of our consolidated financial statements.
Consequently, management is pursuing various financing alternatives to fund our operations so we can continue as a going concern. However, the COVID-19 pandemic has created significant economic uncertainty and volatility in the credit and capital markets. Management plans to secure the necessary financing through potential proceeds from our Purchase Agreement with Lincoln Park and the issue of new equity and/or the entering into of strategic partnership arrangements. However, the ultimate impact of the COVID-19 pandemic on our ability to raise additional capital is unknown and will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration of the COVID-19 outbreak and any new information which may emerge concerning the severity of the COVID-19 pandemic. We may not be able to raise sufficient additional capital and may tailor our drug candidate development program based on the amount of funding we are able to raise in the future. Nevertheless, there is no assurance that these initiatives will be successful.
The consolidated financial statements do not give effect to any adjustments to the amounts and classification of assets and liabilities that may be necessary should we be unable to continue as a going concern. Such adjustments could be material.
Our future funding requirements will depend on many factors, including but not limited to:
Until we can generate a sufficient amount of product revenue to finance our cash requirements, which we may never do, we expect to finance future cash needs primarily through public or private equity offerings, or strategic collaborations. The sale of equity and convertible debt securities may result in dilution to our stockholders and certain of those securities may have rights senior to those of our shares of capital stock. If we raise additional funds through the issuance of preferred stock, convertible debt securities or other debt financing, these securities or other debt could contain covenants that would restrict our operations. Any other third-party funding arrangement could require us to relinquish valuable rights. Economic conditions may affect the availability of funds and activity in equity markets. We do not know whether additional funding will be available on acceptable terms, or at all. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more of our clinical trials or research and development programs or make changes to our operating plan. In addition, we may have to seek a partner for one or more of our product candidates at an earlier stage of development, which would lower the economic value of those programs to us.
Critical Accounting Policies and Estimates
The preparation of financial statements, in conformity with generally accepted accounting principles in the United States, requires companies to establish accounting policies and to make estimates that affect both the amount and timing of the recording of assets, liabilities, revenues and expenses. Some of these estimates require judgments about matters that are inherently uncertain and therefore actual results may differ from those estimates.
A detailed presentation of all of our significant accounting policies and the estimates derived therefrom is included in Note 2 to our consolidated financial statements for the year ended June 30, 2022, contained elsewhere in this Form 10-K. While all of the significant accounting policies are important to our consolidated financial statements, the following accounting policies and the estimates derived therefrom are critical:
64
Fair value of financial instruments
We recognize compensation costs resulting from the issuance of stock-based awards to employees, non-employees and directors as an expense in the statement of operations over the service period based on a measurement of fair value for each stock-based award. Prior to our adoption of Accounting Standards Update 2018-07, Compensation-Stock Compensation (Topic 718), Improvements to Nonemployee Share-Based Payment Accounting (“ASU 2018-07”), stock options granted to non-employee consultants were revalued at the end of each reporting period until vested using the Black-Scholes option-pricing model and the changes in their fair value were recorded as adjustments to expense over the related vesting period. For the years ended June 30, 2022, and 2021, the determination of grant-date fair value for stock option awards was estimated using the Black-Scholes model which includes variables such as the expected volatility of our share price, the anticipated exercise behavior of its grantee, interest rates, and dividend yields. For the years ended June 30, 2022, and 2021, we utilized the plain vanilla method to determine the expected life of stock options. These variables are projected based on our historical data, experience, and other factors. Changes in any of these variables could result in material adjustments to the expense recognized for share-based payments. Such value is recognized as expense over the requisite service period, net of actual forfeitures, using the accelerated attribution method. We recognize forfeitures as they occur. The estimation of stock awards that will ultimately vest requires judgment, and to the extent actual results, or updated estimates, differ from current estimates, such amounts are recorded as a cumulative adjustment in the period estimates are revised.
We have issued warrants for services provided by non-employees. The warrants issued for services have been valued at the fair value of the warrants issued. For the years ended June 30, 2022, and 2021, the determination of grant-date fair value for warrants issued for services was estimated using the Black-Scholes model which includes variables such as the expected volatility of our share price, interest rates, dividend yields, and the term of the warrant. We have also issued shares for services to non-employees which have been valued using the share price of our common stock.
Accruals for research and development expenses and clinical trials
As part of the process of preparing our financial statements, we are required to estimate our expenses resulting from our obligations under contracts with vendors, clinical research organizations and consultants, and under clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment terms that do not match the periods over which materials or services are provided under such contracts. Our objective is to reflect the appropriate expenses in our financial statements by matching those expenses with the period in which services are performed and efforts are expended. We account for these expenses according to the timing of various aspects of the expenses. We determine accrual estimates by taking into account discussion with applicable personnel and outside service providers as to the progress of clinical trials, or the services completed. During the course of a clinical trial, we adjust our clinical expense recognition if actual results differ from our estimates. We make estimates of our accrued expenses as of each balance sheet date based on the facts and circumstances known to us at that time. Our clinical trial accruals are dependent upon the timely and accurate reporting of contract research organizations and other third-party vendors. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in us reporting amounts that are too high or too low for any particular period. For years ended June 30, 2022, and 2021, there were no material adjustments to our prior period estimates of accrued expenses for clinical trials.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Not required for a smaller reporting company.
Item 8. Financial Statements and Supplementary Data
Kintara Therapeutics, Inc.
Consolidated Financial Statements
For the years ended June 30, 2022 and 2021
(expressed in US dollars unless otherwise noted)
65
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of
Kintara Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Kintara Therapeutics, Inc. (the “Company”) as of June 30, 2022 and 2021, the related consolidated statements of operations, stockholders’ equity and cash flows for each of the two years in the period ended June 30, 2022, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of June 30, 2022 and 2021, and the results of its operations and its cash flows for each of the two years in the period ended June 30, 2022, in conformity with accounting principles generally accepted in the United States of America.
Explanatory Paragraph – Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As more fully described in Note 1, the Company has incurred significant losses and needs to raise additional funds to meet its obligations and sustain its operations. These conditions raise substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
66
Accruals for Research and Development Expenses and Clinical Trials
Description of the Matter
As discussed in note 2 to the consolidated financial statements, the Company records accruals for research and development expenses and clinical trials based upon estimates of costs incurred through the balance sheet date that have yet to be invoiced by the contract research organizations (“CRO”) and other third-party vendors.
The Company accounts for these expenses according to the progress of the trial as measured by patient progression and the timing of various aspects of the trial. Estimated accruals are determined based on reviewing contractual terms and through communications with internal clinical personnel and external service providers including CRO’s as to the progress or state of its trials. The principal consideration for our determination that performing procedures related to the clinical trial expenses, specifically related to the year-end accrual for clinical trial costs, is a critical audit matter is that there was judgment by management in determining the progress of the activities included in the individual clinical trial agreements based on internal and external information.
How We Addressed the Matter in Our Audit
To evaluate the accruals for research and development expenses and clinical trials, our audit procedures included, among others, testing the completeness and accuracy of the underlying data used in the estimates and evaluating the significant assumptions including, but not limited, by obtaining an understanding of the Company’s estimation process, corroborating the progress of clinical trials with the Company’s clinical team, and obtaining confirmations directly from third parties.
/s/
Marcum LLP
We have served as the Company’s auditor since 2019.
September 27, 2022
67
Kintara Therapeutics, Inc.
Consolidated Balance Sheets
(In thousands, except par value amounts)
|
|
|
|
June 30, |
|
|
June 30, |
|
||
|
|
Note |
|
$ |
|
|
$ |
|
||
|
|
|
|
|
|
|
|
|
||
Assets |
|
|
|
|
|
|
|
|
||
Current assets |
|
|
|
|
|
|
|
|
||
Cash and cash equivalents |
|
|
|
|
|
|
|
|
||
Prepaid expenses, deposits and other |
|
|
|
|
|
|
|
|
||
Clinical trial deposit |
|
4 |
|
|
— |
|
|
|
|
|
Total current assets |
|
|
|
|
|
|
|
|
||
Clinical trial deposit |
|
4 |
|
|
|
|
|
|
||
Property, equipment and intangibles, net |
|
5 |
|
|
|
|
|
|
||
Total assets |
|
|
|
|
|
|
|
|
||
Liabilities |
|
|
|
|
|
|
|
|
||
Current liabilities |
|
|
|
|
|
|
|
|
||
Accounts payable and accrued liabilities |
|
|
|
|
|
|
|
|
||
Related party payables |
|
6 |
|
|
|
|
|
|
||
Total current liabilities |
|
|
|
|
|
|
|
|
||
Milestone payment liability |
|
3 |
|
|
|
|
|
|
||
Total liabilities |
|
|
|
|
|
|
|
|
||
Stockholders' equity |
|
|
|
|
|
|
|
|
||
Preferred stock |
|
|
|
|
|
|
|
|
||
Authorized |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|||
Issued and outstanding |
|
|
|
|
|
|
|
|
||
|
6,8 |
|
|
|
|
|
|
|||
|
8 |
|
|
|
|
|
|
|||
Common stock |
|
|
|
|
|
|
|
|
||
Authorized |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|||
Issued and outstanding |
|
|
|
|
|
|
|
|
||
|
8 |
|
|
|
|
|
|
|||
Additional paid-in capital |
|
8 |
|
|
|
|
|
|
||
Accumulated deficit |
|
|
|
|
( |
) |
|
|
( |
) |
Accumulated other comprehensive income |
|
|
|
|
|
|
|
|
||
Total stockholders’ equity |
|
|
|
|
|
|
|
|
||
Total liabilities and stockholders’ equity |
|
|
|
|
|
|
|
|
||
Nature of operations, corporate history, going concern and |
|
|
|
|
|
|
|
|
||
Commitments and contingencies (note 10) |
|
|
|
|
|
|
|
|
||
Subsequent events (note 13) |
|
|
|
|
|
|
|
|
||
The accompanying notes are an integral part of these consolidated financial statements.
68
Kintara Therapeutics, Inc.
Consolidated Statements of Operations
(In thousands, except per share amounts)
|
|
|
|
For the years ended June 30, |
|
|||||
|
|
Note |
|
2022 |
|
|
2021 |
|
||
Expenses |
|
|
|
|
|
|
|
|
||
Research and development |
|
|
$ |
|
|
$ |
|
|
||
General and administrative |
|
|
|
|
|
|
|
|
||
Merger costs |
|
3 |
|
|
— |
|
|
|
|
|
In-process research and development |
|
3 |
|
|
— |
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
( |
) |
Other income (loss) |
|
|
|
|
|
|
|
|
||
Foreign exchange |
|
|
|
|
|
|
|
( |
) |
|
Amortization of deferred loan costs |
|
7 |
|
|
— |
|
|
|
( |
) |
Interest, net |
|
7 |
|
|
|
|
|
( | ) |
|
|
|
|
|
|
|
|
|
( |
) |
|
Net loss for the year |
|
|
|
|
( |
) |
|
|
( |
) |
Computation of basic loss per share |
|
|
|
|
|
|
|
|
||
Net loss for the year |
|
|
|
|
( |
) |
|
|
( |
) |
Deemed dividend recognized on beneficial conversion features of Series C Preferred stock issuance |
|
8 |
|
|
— |
|
|
|
( |
) |
Series A Preferred cash dividend |
|
8 |
|
|
( |
) |
|
|
( |
) |
Series B Preferred stock dividend |
|
8 |
|
|
— |
|
|
|
( |
) |
Series C Preferred stock dividend |
|
8 |
|
|
( |
) |
|
|
— |
|
Net loss for the year attributable to common stockholders |
|
|
$ |
|
( |
) |
$ |
|
( |
) |
Basic and fully diluted loss per share |
|
|
|
|
( |
) |
|
|
( |
) |
Basic and fully diluted weighted average number of shares |
|
|
$ |
|
|
$ |
|
|
||
The accompanying notes are an integral part of these consolidated financial statements.
69
Kintara Therapeutics, Inc.
Consolidated Statements of Stockholders’ Equity
For the years ended June 30, 2022 and 2021
(In thousands)
|
|
Number |
|
|
Common |
|
|
Additional |
|
|
Accumulated |
|
|
Preferred |
|
|
Accumulated |
|
|
Total Stockholders' |
|
|||||||
Balance - June 30, 2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||||
Adgero merger (note 3) |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Issuance of Series C Preferred stock |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
||
Series C placement agent warrants |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|
Series C Preferred stock share issuance costs |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
( |
) |
Deemed dividend recognized on beneficial |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
Conversion of Series B Preferred stock to common stock |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
||
Conversion of Series C Preferred stock to common stock |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|||
Series C Agent Warrants exercised |
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
Warrants issued for services |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Exercise of warrants |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Stock options exercised |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Stock option expense |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Series A Preferred cash dividend |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Series B Preferred stock dividend |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
||
Net loss for the year |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance - June 30, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||||
Issuance of shares and warrants - net of issue costs |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||||
Conversion of Series C Preferred stock to common stock |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|
|
— |
|
|||
Warrants issued for services |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Exercise of 2020 Investor Warrants for cash |
|
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
Exercise of pre-funded warrants for cash |
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|||
Stock option expense |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
||
Series A Preferred cash dividend |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Series C Preferred stock dividend |
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
— |
|
|||
Net loss for the year |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
( |
) |
|
|
( |
) |
Balance - June 30, 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
|
|
||||||
The accompanying notes are an integral part of these consolidated financial statements.
70
Kintara Therapeutics, Inc.
Consolidated Statements of Cash Flows
June 30, 2022
(In thousands)
|
|
|
|
For the years ended June 30, |
|
|||||
|
|
|
|
2022 |
|
|
2021 |
|
||
|
|
Note |
|
$ |
|
|
$ |
|
||
Cash flows from operating activities |
|
|
|
|
|
|
|
|
||
Net loss for the year |
|
|
|
|
( |
) |
|
|
( |
) |
Adjustments to reconcile net loss to net cash used in operating |
|
|
|
|
|
|
|
|
||
Amortization of intangible assets |
|
|
|
|
— |
|
|
|
|
|
Depreciation of property and equipment |
|
5 |
|
|
|
|
|
|
||
Impairment of in-process research and development |
|
3 |
|
|
— |
|
|
|
|
|
Change in fair value of milestone liability |
|
3 |
|
|
( |
) |
|
|
( |
) |
Interest expense |
|
7 |
|
|
— |
|
|
|
|
|
Amortization of deferred loan costs |
|
7 |
|
|
— |
|
|
|
|
|
Warrants issued for services |
|
8 |
|
|
|
|
|
|
||
Stock option expense |
|
8 |
|
|
|
|
|
|
||
Changes in operating assets and liabilities |
|
|
|
|
|
|
|
|
||
Prepaid expenses, deposits and other |
|
|
|
|
( | ) |
|
|
( | ) |
Clinical trial deposits |
|
4 |
|
|
( |
) |
|
|
( |
) |
Accounts payable and accrued liabilities |
|
|
|
|
|
|
|
( |
) |
|
Related party payables |
|
|
|
|
|
|
|
( |
) |
|
Net cash used in operating activities |
|
|
|
|
( |
) |
|
|
( |
) |
Cash flows from investing activities |
|
|
|
|
|
|
|
|
||
Cash acquired on merger with Adgero |
|
3 |
|
|
— |
|
|
|
|
|
Purchase of equipment |
|
|
|
|
— |
|
|
|
( |
) |
Proceeds on sale of equipment |
|
|
|
|
— |
|
|
|
|
|
Net cash provided by investing activities |
|
|
|
|
— |
|
|
|
|
|
Cash flows from financing activities |
|
|
|
|
|
|
|
|
||
Net proceeds from the issuance of shares and warrants |
|
8 |
|
|
|
|
|
|
||
Warrants exercised for cash |
|
8 |
|
|
|
|
|
|
||
Stock options exercised for cash |
|
|
|
|
— |
|
|
|
|
|
Proceeds from loan |
|
7 |
|
|
— |
|
|
|
|
|
Repayment of loan |
|
7 |
|
|
— |
|
|
|
( |
) |
Interest paid |
|
7 |
|
|
— |
|
|
|
( |
) |
Series A preferred cash dividend |
|
8 |
|
|
( |
) |
|
|
( |
) |
Net cash provided by financing activities |
|
|
|
|
|
|
|
|
||
Increase in cash and cash equivalents |
|
|
|
|
|
|
|
|
||
Cash and cash equivalents – beginning of year |
|
|
|
|
|
|
|
|
||
Cash and cash equivalents – end of year |
|
|
|
|
|
|
|
|
||
Supplementary information (note 11) |
|
|
|
|
|
|
|
|
||
The accompanying notes are an integral part of these consolidated financial statements.
71
Kintara Therapeutics, Inc.
Notes to Consolidated Financial Statements
June 30, 2022
(In thousands)
Nature of operations
Kintara Therapeutics, Inc. (the “Company”) is a clinical-stage drug development company with a focus on the development of novel cancer therapies for patients with unmet medical needs. The Company is developing two late-stage, Phase 3-ready therapeutics – VAL-083 for glioblastoma multiforme and REM-001 for cutaneous metastatic breast cancer. In order to accelerate the Company’s development timelines, it leverages existing preclinical and clinical data from a wide range of sources. The Company may seek marketing partnerships in order to potentially offset clinical costs and to generate future royalty revenue from approved indications of its product candidates.
Corporate history
The Company is a Nevada corporation formed on June 24, 2009, under the name Berry Only, Inc. On January 25, 2013, the Company entered into and closed an exchange agreement (the “Exchange Agreement”), with Del Mar Pharmaceuticals (BC) Ltd. (“Del Mar (BC)”), 0959454 B.C. Ltd. (“Callco”), and 0959456 B.C. Ltd. (“Exchangeco”) and the security holders of Del Mar (BC). Upon completion of the Exchange Agreement, Del Mar (BC) became a wholly-owned subsidiary of the Company (the “Reverse Acquisition”).
On June 9, 2020, the Company entered into an Agreement and Plan of Merger and Reorganization (the “Merger Agreement”), by and among Adgero Acquisition Corp., the Company’s wholly-owned subsidiary incorporated in the State of Delaware (“Merger Sub”), and Adgero Biopharmaceuticals Holdings, Inc., a Delaware corporation (“Adgero”). On August 19, 2020, upon the terms and subject to the conditions set forth in the Merger Agreement, Merger Sub merged with and into Adgero (the “Merger”), the separate corporate existence of Merger Sub ceased and Adgero continued its existence under Delaware law as the surviving corporation in the Merger and became a direct, wholly-owned subsidiary of the Company. As a result of the Merger, each issued and outstanding share of Adgero common stock, par value $
Following the completion of the Merger, the Company changed its name from DelMar Pharmaceuticals, Inc. to Kintara Therapeutics, Inc. and began trading on The Nasdaq Capital Market LLC under the symbol “KTRA”.
Kintara Therapeutics, Inc. is the parent company of Del Mar (BC), a British Columbia, Canada corporation and Adgero, a Delaware corporation, which are clinical stage companies with a focus on the development of drugs for the treatment of cancer. The Company is also the parent company to Callco and Exchangeco which are British Columbia, Canada corporations. Callco and Exchangeco were formed to facilitate the Reverse Acquisition. In connection with the Adgero merger, the Company also became the parent company of Adgero Biopharmaceuticals, Inc. (“Adgero Bio”), formerly a wholly-owned subsidiary of Adgero.
References to the Company refer to the Company and its wholly-owned subsidiaries.
Going concern and management plans
These consolidated financial statements have been prepared on a going concern basis, which assumes that the Company will continue its operations for the foreseeable future and contemplates the realization of assets and the settlement of liabilities in the normal course of business.
For the year ended June 30, 2022, the Company reported a loss of $
72
Consequently, management is pursuing various financing alternatives to fund the Company’s operations so it can continue as a going concern. However, the coronavirus (“COVID-19”) pandemic has created significant economic uncertainty and volatility in the credit and capital markets. Management plans to continue to pursue opportunities to secure the necessary financing through the issue of new equity, debt, and/or the entering into of strategic partnership arrangements but the ultimate impact of the COVID-19 pandemic on the Company’s ability to raise additional capital is unknown and will depend on future developments, which are highly uncertain and cannot be predicted with confidence, including the duration of the COVID-19 outbreak and any new information which may emerge concerning the severity of the COVID-19 pandemic. The Company may not be able to raise sufficient additional capital and may tailor its drug candidate development programs based on the amount of funding the Company is able to raise in the future. Nevertheless, there is no assurance that these initiatives will be successful.
These consolidated financial statements do not give effect to any adjustments to the amounts and classification of assets and liabilities that may be necessary should the Company be unable to continue as a going concern. Such adjustments could be material.
Basis of presentation
The consolidated financial statements of the Company have been prepared in accordance with United States Generally Accepted Accounting Principles (“U.S. GAAP”) and are presented in United States dollars. The functional currency of the Company and each of its subsidiaries is the United States dollar.
The principal accounting policies applied in the preparation of these consolidated financial statements are set out below and have been consistently applied to all years presented.
Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries, Adgero, Adgero Bio, Del Mar BC, Callco, and Exchangeco as of, and for the years ended June 30, 2022, and 2021. All intercompany balances and transactions have been eliminated in consolidation.
Use of estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions about future events that affect the reported amounts of assets, liabilities, expenses, contingent assets, and contingent liabilities as at the end of, or during, the reporting period. Actual results could significantly differ from those estimates. Significant areas requiring management to make estimates include the fair value of the milestone payment liability, the valuation of equity instruments issued for services, clinical trial accruals, deferred tax valuation allowance and assessment of going concern. Further details of the nature of these assumptions and conditions may be found in the relevant notes to these consolidated financial statements.
Cash and cash equivalents
Cash and cash equivalents consist of cash and highly liquid investments with original maturities from the purchase date of three months or less that can be readily convertible into known amounts of cash. Cash and cash equivalents are held at recognized Canadian and United States financial institutions. Interest earned is recognized in the consolidated statement of operations.
Foreign currency translation
The functional currency of the Company at June 30, 2022, is the United States dollar. Transactions that are denominated in a foreign currency are remeasured into the functional currency at the current exchange rate on the date of the transaction. Any foreign-currency denominated monetary assets and liabilities are subsequently remeasured at current exchange rates, with gains or losses recognized as foreign exchange losses or gains in the consolidated statement of operations. Non-monetary assets and liabilities are translated at historical exchange rates. Expenses are translated at average exchange rates during the period. Exchange gains and losses are included in consolidated statement of operations for the period.
Acquired in-process research and development expense
The Company acquired in-process research and development assets in connection with its Merger with Adgero (note 3). As the acquired in-process research and development assets were deemed to have no current or alternative future use, an expense of $
73
Property and equipment
Property and equipment is stated at cost less accumulated depreciation. Depreciation is calculated on a straight-line basis over its estimated useful life of
Income taxes
The Company uses the asset and liability method of accounting for income taxes. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to the differences between the consolidated financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. To the extent that deferred tax assets cannot be recognized under the preceding criteria, the Company establishes valuation allowances, as necessary, to reduce deferred tax assets to the amounts expected to be realized.
As of June 30, 2022, and 2021, all deferred tax assets were fully offset by a valuation allowance. The realization of deferred tax assets is dependent upon future federal, state and foreign taxable income. The Company’s judgments regarding deferred tax assets may change due to future market conditions, as the Company expands into international jurisdictions, due to changes in U.S. or international tax laws and other factors.
These changes, if any, may require material adjustments to the Company’s deferred tax assets, resulting in a reduction in net income or an increase in net loss in the period in which such determinations are made. The Company recognizes the impact of uncertain tax positions based upon a two-step process. To the extent that a tax position does not meet a more-likely-than-not level of certainty, no impact is recognized in the consolidated financial statements. If a tax position meets the more-likely-than-not level of certainty, it is recognized in the consolidated financial statements at the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The Company’s policy is to analyze the Company’s tax positions taken with respect to all applicable income tax issues for all open tax years in each respective jurisdiction. Interest and penalties with respect to uncertain tax positions would be included in income tax expense. As of June 30, 2022, the Company concluded that there were
The Company does not record U.S. income taxes on the undistributed earnings of its foreign subsidiaries based upon the Company’s intention to permanently reinvest undistributed earnings to ensure sufficient working capital and further expansion of existing operations outside the United States. As June 30, 2022, the Company’s foreign subsidiaries operated at a cumulative deficit for U.S. earnings and profit purposes. In the event the Company is required to repatriate funds from outside of the United States, such repatriation would be subject to local laws, customs, and tax consequences. Determination of the amount of unrecognized deferred tax liability related to these earnings is not practicable.
Financial instruments
The Company has financial instruments that are measured at fair value. To determine the fair value, the Company uses the fair value hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs market participants would use to value an asset or liability and are developed based on market data obtained from independent sources. Unobservable inputs are inputs based on assumptions about the factors market participants would use to value an asset or liability. The three levels of inputs that may be used to measure fair value are as follows:
Assets and liabilities are classified based on the lowest level of input that is significant to the fair value measurements. Changes in the observability of valuation inputs may result in a reclassification of levels for certain securities within the fair value hierarchy. As of June 30, 2022, the Company’s milestone payment liability was measured using level 3 inputs (note 3).
74
The Company’s financial instruments consist of cash and cash equivalents, other receivables, accounts payable, and related party payables. The carrying values of cash and cash equivalents, other receivables, accounts payable and related party payables approximate their fair values due to the immediate or short-term maturity of these financial instruments.
Intangible assets
Patents
Expenditures associated with the filing, or maintenance of patents, licensing or technology agreements are expensed as incurred. Costs previously recognized as an expense are not recognized as an asset in subsequent periods. If the Company achieves regulatory approval, patent costs will be deferred and amortized over the remaining life of the related patent.
Accruals for research and development expenses and clinical trials
As part of the process of preparing its financial statements, the Company is required to estimate its expenses resulting from its obligations under contracts with vendors, clinical research organizations and consultants, and under clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment terms that do not match the periods over which materials or services are provided under such contracts. The Company’s objective is to reflect the appropriate expenses in its financial statements by matching those expenses with the period in which services are performed and efforts are expended. The Company accounts for these expenses according to the timing of various aspects of the expenses. The Company determines accrual estimates by taking into account discussion with applicable personnel and outside service providers as to the progress of clinical trials, or the services completed. During the course of a clinical trial, the Company adjusts its clinical expense recognition if actual results differ from its estimates. The Company makes estimates of its accrued expenses as of each balance sheet date based on the facts and circumstances known to it at that time. The Company’s clinical trial accruals are dependent upon the timely and accurate reporting of contract research organizations and other third-party vendors. Although the Company does not expect its estimates to be materially different from amounts actually incurred, its understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in it reporting amounts that are too high or too low for any particular period. For the years ended June 30, 2022, and 2021, there were no material adjustments to the Company’s prior period estimates of accrued expenses for clinical trials.
Warrants and shares issued for services
The Company has issued equity instruments for services provided by employees and non-employees. The equity instruments are valued at the fair value of the instrument issued.
Stock options
The Company recognizes compensation costs resulting from the issuance of stock-based awards to employees, non-employees and directors as an expense in the statement of operations over the service period based on a measurement of fair value for each stock-based award. Prior to our adoption of Accounting Standards Update ("ASU") 2018-07, Compensation-Stock Compensation (Topic 718), Improvements to Nonemployee Share-Based Payment Accounting (“ASU 2018-07”), stock options granted to non-employee consultants were revalued at the end of each reporting period until vested using the Black-Scholes option-pricing model and the changes in their fair value were recorded as adjustments to expense over the related vesting period. For the years ended June 30, 2022, and 2021, the determination of grant-date fair value for stock option awards was estimated using the Black-Scholes model, which includes variables such as the expected volatility of our share price, the anticipated exercise behavior of its grantee, interest rates, and dividend yields. For years ended June 30, 2022, and 2021, the Company utilized the plain vanilla method to determine the expected life of stock options. These variables are projected based on our historical data, experience, and other factors. Changes in any of these variables could result in material adjustments to the expense recognized for share-based payments. Such value is recognized as expense over the requisite service period, net of actual forfeitures, using the accelerated attribution method. The Company recognizes forfeitures as they occur. The estimation of stock awards that will ultimately vest requires judgment, and to the extent actual results, or updated estimates, differ from current estimates, such amounts are recorded as a cumulative adjustment in the period estimates are revised.
Loss per share
Income or loss per share is calculated based on the weighted average number of common shares outstanding. For the years ended June 30, 2022, and 2021 diluted loss per share does not differ from basic loss per share since the effect of the Company’s warrants, stock options, performance stock units, and convertible preferred shares is anti-dilutive. As of June 30, 2022, potential common shares of
75
Segment information
The Company identifies its operating segments based on business activities, management responsibility and geographical location. The Company operates within a operating segment being the research and development of cancer indications, and operates primarily in
Recent accounting pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (“FASB”) or other standard setting bodies that are adopted by the Company as of the specified effective date.
Recently adopted
ASU 2020-06 — Debt - Debt with conversion and other options (subtopic 470-20) and derivatives and hedging – contracts in entity’s own equity (subtopic 815-40): accounting for convertible instruments and contracts in an entity’s own equity
The amendments in this update are intended to simplify the accounting for certain financial instruments with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own equity. The ASU is part of the FASB’s simplification initiative, which aims to reduce unnecessary complexity in U.S. GAAP. For public business entities that are not smaller reporting companies, the ASU’s amendments are effective for fiscal years beginning after December 15, 2021, and interim periods within those fiscal years. For all other entities, the effective date is for fiscal years beginning after December 15, 2023, and interim periods within those fiscal years. The guidance may be early adopted for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years. The Company adopted ASU 2020-06 effective July 1, 2021. The adoption of ASU 2020-06 did not have a material impact on the Company’s financial statements.
During the year ended June 30, 2022, there have been no new, or existing, recently issued accounting pronouncements that are of significance, or potential significance, that impact the Company’s consolidated financial statements.
On June 9, 2020, the Company, Adgero Acquisition Corp., a wholly-owned subsidiary of the Company (“Merger Sub”), and Adgero, entered into an Agreement and Plan of Merger and Reorganization (the “Merger Agreement”) pursuant to which upon closing the Merger Sub will merge with and into Adgero, with Adgero surviving the merger and becoming a direct, wholly-owned subsidiary of the Company (the “Merger”).
Subject to the terms and conditions of the Merger Agreement, at the effective time of the Merger (the “Effective Time”), (i) each outstanding share of Adgero common stock (the “Adgero Common Stock”) was converted into shares of Company common stock (the “Kintara Common Stock”) based on the exchange ratio, (ii) each outstanding warrant to purchase Adgero Common Stock was converted into a warrant exercisable for that number of shares of Kintara Common Stock equal to the product of (x) the aggregate number of shares of Adgero Common Stock for which such warrant was exercisable and (y) the Exchange Ratio (as defined in the Merger Agreement); and (iii) each outstanding Adgero stock option, whether vested or unvested, that had not been exercised was cancelled for no consideration. On August 19, 2020, upon the terms and subject to the conditions set forth in the Merger Agreement, Merger Sub merged with and into Adgero, the separate corporate existence of Merger Sub ceased and Adgero continued its existence under Delaware law as the surviving corporation and a direct, wholly-owned subsidiary of the Company.
The Exchange Ratio in the Merger Agreement was negotiated so that the existing stockholders of Adgero would own
Under the terms of the Merger Agreement, upon closing of the Merger, the Company issued
76
In connection with the Merger, the Company completed a private placement of Series C Convertible Preferred Stock in three separate closings (note 8).
To determine the accounting for this transaction under ASU 2017-01, an assessment was made as to whether an integrated set of assets and activities should be accounted for as an acquisition of a business or an asset acquisition. The guidance requires an initial screen test to determine if substantially all of the fair value of the gross assets acquired is concentrated in a single asset or group of similar assets. If that screen is met, the set is not a business. In connection with the Merger, substantially all of the fair value was concentrated in in-process research and development (“IPR&D”). As such, the Merger has been treated as an acquisition of Adgero assets and an assumption of Adgero liabilities.
The Company incurred approximately $
The following summarizes total consideration transferred to the Adgero stockholders under the Merger as well as the assets acquired and liabilities assumed under the Merger:
|
|
$ |
|
|
Consideration: |
|
|
|
|
Common stock |
|
|
|
|
Warrants |
|
|
|
|
Success fee shares |
|
|
|
|
|
|
|
|
|
Net assets acquired: |
|
|
|
|
Cash |
|
|
( |
) |
Other current assets |
|
|
( |
) |
Property and equipment (note 5) |
|
|
( |
) |
Accounts payable and accrued liabilities |
|
|
|
|
Milestone payment liability |
|
|
|
|
In-process research and development |
|
|
|
|
The fair value of the IPR&D assets has been expensed as a charge in the consolidated statements of operations for the year ended June 30, 2021, as there is no alternative use for these assets. Property and equipment includes office furniture that was subsequently sold and laboratory equipment that was put into use during the third quarter of the year ended June 30, 2021.
The milestone payment liability relates to an asset purchase agreement with St. Cloud Investments, LLC (“St. Cloud”) that Adgero has regarding the acquisition of REM-001. The Agreement, as amended, is dated November 26, 2012 (the “St. Cloud Agreement”). Pursuant to the terms of the St. Cloud Agreement, the Company is obligated to make certain payments under the agreement. The future contingent amounts payable under that agreement are as follows:
With respect to the $300 and $700 potential milestone payments referenced above (each a “Milestone Payment”), if either such Milestone Payment becomes payable, and in the event the Company elects to pay either such Milestone Payment in shares of its common stock, the value of the common stock will equal the average of the closing price per share of the Company’s common stock over the twenty (20) trading days following the first public announcement of the applicable event described above.
The milestone payment liability has been estimated using a scenario-based method (or “SBM”). An SBM is an income-based approach under which possible outcomes are identified, the contingent consideration payoff of each outcome is probability weighted, and then a suitable discount rate is used to arrive at the expected present value of the contingent consideration at the valuation date. The probability used in the valuation was based on published research for the probability of success of oncology companies at a similar stage of development as the Company.
77
based on an estimate of the planned timing of completion of the respective development achievement that would result in payment of the respective milestones.
|
|
$ |
|
|
Balance – June 30, 2020 |
|
|
— |
|
Addition |
|
|
|
|
Change in fair value estimate |
|
|
( |
) |
Balance – June 30, 2021 |
|
|
|
|
Change in fair value estimate |
|
|
( |
) |
Balance – June 30, 2022 |
|
|
|
|
The Company has entered into an agreement with a contract research organization (“CRO”) for the management of the Company’s registration study for glioblastoma multiforme. Under the agreement, the Company will supply the drug for the study and the CRO will manage all operational aspects of the study including site activation and patient enrollment. The Company is required to make certain payments under the agreement related to patient enrollment milestones. For the year ended June 30, 2022, the Company has recognized $
In relation to this study, the Company has made a deposit payment of $
|
|
$ |
|
|
Balance, June 30, 2020 |
|
|
|
|
Acquired in Adgero merger (note 3) |
|
|
|
|
Laboratory equipment purchased |
|
|
|
|
Disposal of furniture |
|
|
( |
) |
Less depreciation and amortization |
|
|
( |
) |
Balance, June 30, 2021 |
|
|
|
|
Less depreciation |
|
|
( |
) |
Balance, June 30, 2022 |
|
|
|
|
At June 30, 2022, the total capitalized cost of intangibles was $
Valent Technologies, LLC Agreements
One of the Company’s officers is a principal of Valent Technologies, LLC (“Valent”) and as result Valent is a related party to the Company.
On September 12, 2010, the Company entered into a Patent Assignment Agreement (the “Valent Assignment Agreement”) with Valent pursuant to which Valent transferred to the Company all its right, title and interest in and to the patents for VAL-083 owned by Valent. The Company now owns all rights and title to VAL-083 and is responsible for the drug’s further development and commercialization. In accordance with the terms of the Valent Assignment Agreement, Valent is entitled to receive a future royalty on all revenues derived from the development and commercialization of VAL-083. In the event that the Company terminates the agreement, the Company may be entitled to receive royalties from Valent’s subsequent development of VAL-083 depending on the development milestones the Company has achieved prior to the termination of the Valent Assignment Agreement.
On September 30, 2014, the Company entered into an exchange agreement (the “Valent Exchange Agreement”) with Valent and Del Mar (BC). Pursuant to the Valent Exchange Agreement, Valent exchanged its loan payable in the outstanding amount of $
78
Company’s Series A Preferred Stock. The Series A Preferred Stock has a stated value of $
Related party payables
At June 30, 2022 there is an aggregate amount of $
|
|
$ |
|
|
Balance – June 30, 2020 |
|
|
— |
|
Funding |
|
|
|
|
Financing costs |
|
|
( |
) |
Interest expense |
|
|
|
|
Amortization of deferred financing costs |
|
|
|
|
Payment of principal and interest |
|
|
( |
) |
Balance – June 30, 2021 |
|
|
— |
|
During the year ended June 30, 2021, the Company received a loan of $
The NBTS Warrants were valued at $
Preferred stock
Series C Preferred Stock
|
|
Series C Preferred Stock |
|
|||||
|
|
Number |
|
|
$ |
|
||
Balance – June 30, 2020 |
|
|
— |
|
|
|
— |
|
Issuance |
|
|
|
|
|
|
||
Issued on exercise of Series C Agent Warrants |
|
|
|
|
|
|
||
Conversion of Series C Preferred stock to common stock |
|
|
( |
) |
|
|
( |
) |
Balance – June 30, 2021 |
|
|
|
|
|
|
||
Conversion of Series C Preferred stock to common stock |
|
|
( |
) |
|
|
( |
) |
Balance – June 30, 2022 |
|
|
|
|
|
|
||
In connection with the Merger (note 3), in August 2020, the Company issued
79
the Series C Preferred Stock, on the 12th, 24th, 36th and 48th month, anniversary of the initial closing of the private placement which occurred on August 19, 2020.
The Series C Preferred Stock dividends do not require declaration by the board of directors and are accrued annually as of the date the dividend is earned in an amount equal to the fair value of the Company’s common stock on the dates the respective dividends are paid. The fair value of the Series C Preferred Stock dividend paid on August 19, 2021, was determined by multiplying the dividends paid of
Total gross proceeds from the private placement were $
The Company’s Series C Preferred Stock outstanding, conversion shares, and future dividends as of June 30, 2022, are as follows:
Series |
|
Number |
|
|
Conversion Price |
|
|
Number of conversion shares |
|
|
Dividend Shares |
|
||||
Series 1 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Series 2 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Series 3 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Series C Dividends |
|
Dividend Shares |
|
|
10% - August 19, 2021 (actual) |
|
|
|
|
15% - August 19, 2022 (estimated) |
|
|
|
|
20% - August 19, 2023 (estimated) |
|
|
|
|
25% - August 19, 2024 (estimated) |
|
|
|
|
|
|
|
|
|
The conversion feature of the Series C Convertible Preferred Stock at the time of issuance was determined to be beneficial on the commitment date. Because the Series C Convertible Preferred Stock was perpetual with no stated maturity date, and the conversions could occur any time from inception, the Company immediately recorded a non-cash deemed dividend of $
The Series C Preferred Stock shall with respect to distributions of assets and rights upon the occurrence of a liquidation, rank (i) senior to the Company’s common stock and (ii) senior to any other class or series of capital stock of the Company hereafter created which does not expressly rank pari passu with, or senior to, the Series C Preferred Stock. The Series C Preferred Stock shall be pari passu in liquidation to the Company’s Series A Preferred Stock. The liquidation value of the Series C Preferred Stock at June 30, 2022, is the stated value of $
Series B Preferred Stock
|
|
Series B Preferred Stock |
|
|||||
|
|
Number |
|
|
$ |
|
||
Balance – June 30, 2020 |
|
|
|
|
|
|
||
Conversion of Series B Preferred stock to common stock |
|
|
( |
) |
|
|
( |
) |
Balance – June 30, 2021 and 2022 |
|
|
— |
|
|
|
— |
|
During the year ended June 30, 2016, the Company issued
80
the Series B Preferred Stock were entitled to an annual cumulative, in arrears, dividend at the rate of
In addition, the Company and the Series B Preferred Stockholders entered into a royalty agreement, pursuant to which the Company will pay the holders of the Series B Preferred Stock, in aggregate, a single-digit royalty based on their pro rata ownership of the Series B Preferred Stock on products sold directly by the Company or sold pursuant to a licensing or partnering arrangement.
Series A Preferred Stock
Effective September 30, 2014, the Company filed a Certificate of Designation of Series A Preferred Stock (the “Series A Certificate of Designation”) with the Secretary of State of Nevada. Pursuant to the Series A Certificate of Designation, the Company designated
The Series A Preferred Stock shall with respect to distributions of assets and rights upon the occurrence of a liquidation, rank (i) senior to the Company’s common stock, and (ii) senior to any other class or series of capital stock of the Company hereafter created which does not expressly rank pari passu with, or senior to, the Series A Preferred Stock. The Series A Preferred Stock shall be pari passu in liquidation to the Company’s Series C Preferred Stock. The liquidation value of the Series A Preferred stock at June 30, 2022, of $
There was
81
Common stock
Amended articles of incorporation
On June 25, 2021, the Company amended its articles of incorporation to increase the number of authorized shares of common stock from
Stock issuances
Year ended June 30, 2022
Registered direct financing - September 28, 2021
On September 28, 2021,
The net proceeds from the September Offering were $
The 2022 Investor Warrants are exercisable at $1.25 per share until their expiry on
As of June 30, 2022, all of the
Registered direct financing - April 14, 2022
On April 14, 2022,
The net proceeds from the April Offering were approximately $
The 2022 April Investor Warrants are exercisable at $0.41 per share until their expiry on
82
Stock options
2017 Omnibus Incentive Plan
As subsequently approved by the Company’s stockholders at an annual meeting of stockholders, on April 11, 2018, the Company’s board of directors approved the adoption of the Company’s 2017 Omnibus Equity Incentive Plan (the “2017 Plan”), as amended. The board of directors also approved a form of Performance Stock Unit Award Agreement to be used in connection with grants of performance stock units (“PSUs”) under the 2017 Plan. As approved by the Company’s stockholders on June 21, 2022, the number of common shares available under the 2017 Plan is
The maximum number of shares of Company common stock with respect to which any one participant may be granted awards during any calendar year is
During the year ended June 30, 2022, a total of
In addition, 2,715 stock options previously issued to an officer of the Company were modified such that 754 stock options that were to vest over the period December 15, 2022, to September 15, 2023, were changed to vest on a contingent basis dependent on the achievement of certain strategic partnership initiatives. As the conditions to meet the contingent vesting were unlikely to be met, the compensation expense for these stock options was recognized over the pre-amendment basis. The contingent vesting stock options were subsequently forfeited upon the termination of that officer. In relation to the termination of two officers of the Company, the Company has recognized $120 in stock option expense due to the acceleration of vesting of certain stock options granted to those officers.
The following table sets forth changes in stock options outstanding under all plans:
|
|
Number of |
|
|
Weighted |
|
||
Balance – June 30, 2020 |
|
|
|
|
|
|
||
Granted |
|
|
|
|
|
|
||
Exercised |
|
|
( |
) |
|
|
|
|
Forfeited |
|
|
( |
) |
|
|
|
|
Expired |
|
|
( |
) |
|
|
|
|
Balance – June 30, 2021 |
|
|
|
|
|
|
||
Granted |
|
|
|
|
|
|
||
Expired |
|
|
( |
) |
|
|
|
|
Forfeited |
|
|
( |
) |
|
|
|
|
Balance – June 30, 2022 |
|
|
|
|
|
|
||
83
The following table summarizes stock options outstanding and exercisable under all plans at June 30, 2022:
Exercise price |
|
|
Number |
|
|
Weighted |
|
|
Number |
|
||||
|
|
|
|
|
|
|
|
|
|
— |
|
|||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
— |
|
|||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
||||
Stock options issued during the years ended June 30, 2022, and 2021, have been valued using a Black-Scholes pricing model with the following assumptions:
|
|
June 30, |
|
|
June 30, |
|
||
Dividend rate |
|
|
— |
% |
|
|
— |
% |
Volatility |
|
|
|
|
||||
Risk-free rate |
|
|
|
|
||||
Term – years |
|
|
|
|
||||
The estimated volatility of the Company’s common stock at the date of issuance of the stock options is based on the historical volatility of the Company. The risk-free interest rate is based on rates published by the government for bonds with a maturity similar to the expected remaining life of the stock options at the valuation date. The expected life of the stock options has been estimated using the plain vanilla method.
84
The Company has recognized the following amounts as stock option expense for the periods noted:
|
|
Years ended June 30, |
|
|||||
|
|
2022 |
|
|
2021 |
|
||
Research and development |
|
|
|
|
|
|
||
General and administrative |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
All of the stock option expense for the periods ended June 30, 2022, and 2021, has been recognized as additional paid in capital. The aggregate intrinsic value of stock options outstanding at June 30, 2022, was
The following table sets forth changes in unvested stock options under all plans:
|
|
Number of |
|
|
Weighted |
|
||
Unvested at June 30, 2020 |
|
|
|
|
|
|
||
Granted |
|
|
|
|
|
|
||
Vested |
|
|
( |
) |
|
|
|
|
Forfeited |
|
|
( |
) |
|
|
|
|
Unvested at June 30, 2021 |
|
|
|
|
|
|
||
Granted |
|
|
|
|
|
|
||
Vested |
|
|
( |
) |
|
|
|
|
Forfeited |
|
|
( |
) |
|
|
|
|
Unvested at June 30, 2022 |
|
|
|
|
|
|
||
The aggregate intrinsic value of unvested stock options at June 30, 2022 was
Common stock warrants
The following table sets forth changes in outstanding warrants:
|
|
Number of |
|
|
Weighted average exercise price |
|
||
Balance – June 30, 2020 |
|
|
|
|
|
|
||
Issuance of Adgero Warrants |
|
|
|
|
|
|
||
Exercise of warrants (ii) |
|
|
( |
) |
|
|
|
|
Warrants issued for services (i) |
|
|
|
|
|
|
||
Expiry of warrants |
|
|
( |
) |
|
|
|
|
Balance – June 30, 2021 |
|
|
|
|
|
|
||
Issuance of 2022 Investor Warrants |
|
|
|
|
|
|
||
Issuance of PFW |
|
|
|
|
|
|
||
Issuance of 2022 Agent Warrants |
|
|
|
|
|
|
||
Issuance of 2022 April Investor Warrants |
|
|
|
|
|
|
||
Issuance of 2022 April Agent Warrants |
|
|
|
|
|
|
||
Exercise of PFW |
|
|
( |
) |
|
|
|
|
Exercise of 2020 Investor Warrants |
|
|
( |
) |
|
|
|
|
Expiry of warrants (iii) |
|
|
( |
) |
|
|
|
|
Balance – June 30, 2022 |
|
|
|
|
|
|
||
85
The following table summarizes the Company’s outstanding warrants as of June 30, 2022:
Description of warrants |
|
Number |
|
|
Exercise |
|
|
Expiry date |
||
2022 April Investor warrants |
|
|
|
|
|
|
|
|||
2022 Investor warrants |
|
|
|
|
|
|
|
|||
2020 Investor warrants |
|
|
|
|
|
|
|
|||
2019 Investor warrants |
|
|
|
|
|
|
|
|||
2018 Investor warrants |
|
|
|
|
|
|
|
|||
NBTS Warrants |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
Warrants issued for services |
|
|
|
|
|
|
|
|||
2022 April Agent warrants |
|
|
|
|
|
|
|
|||
2022 Agent warrants |
|
|
|
|
|
|
|
|||
2019 Agent warrants |
|
|
|
|
|
|
|
|||
2018 Agent warrants |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
||
Series C preferred stock warrants
In connection with the Series C Preferred Stock private placement, the Company initially issued
86
The Series C Agent Warrants were valued at a total of $
The following table sets forth changes in outstanding Series C Agent Warrants:
|
|
Balance, |
|
|
Number of |
|
|
Number of |
|
|
Balance, |
|
|
Exercise |
|
|||||
Issuance of Preferred Series C-1 Agent Warrants |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Issuance of Preferred Series C-2 Agent Warrants |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Issuance of Preferred Series C-3 Agent Warrants |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
The following table summarizes the Company’s outstanding Series C Agent Warrants as of June 30, 2022:
Series C Agent Warrants |
|
Number |
|
|
Conversion |
|
|
Number of |
|
|
Cumulative |
|
||||
Series 1 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Series 2 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Series 3 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
For the years ended June 30, 2022, and 2021, the Company did not record a provision for income taxes due to a full valuation allowance against the deferred tax assets.
Significant components of the Company’s deferred tax assets and deferred tax liabilities are shown below:
|
|
June 30, |
|
|
June 30, |
|
||
Deferred tax assets: |
|
|
|
|
|
|
||
Non-capital losses carried forward |
|
|
|
|
|
|
||
Stock-based compensation |
|
|
|
|
|
|
||
Capital losses carried forward |
|
|
|
|
|
|
||
Financing costs |
|
|
|
|
|
|
||
Bonus - compensation |
|
|
|
|
|
— |
|
|
Scientific research and development |
|
|
|
|
|
|
||
Scientific research and development – Investment |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
||
Deferred tax liabilities: |
|
|
|
|
|
|
||
Scientific research and development – ITC |
|
|
( |
) |
|
|
( |
) |
|
|
|
|
|
|
|
||
Valuation allowance |
|
|
( |
) |
|
|
( |
) |
Net future tax assets |
|
|
— |
|
|
|
— |
|
The income tax benefit of these tax attributes has not been recorded in these consolidated financial statements because of the uncertainty of their recovery. The Company’s effective income tax rate differs from the statutory income tax rate of
87
The differences arise from the following items:
|
|
June 30, |
|
|
June 30, |
|
||
Tax recovery at statutory income tax rates |
|
|
( |
) |
|
|
( |
) |
Permanent differences |
|
|
|
|
|
|
||
Effect of rate differentials between jurisdictions |
|
|
( |
) |
|
|
( |
) |
Effect of foreign exchange rates |
|
|
|
|
|
( |
) |
|
Non-capital losses acquired with Adgero |
|
|
— |
|
|
|
( |
) |
Scientific research and development – ITC |
|
|
( |
) |
|
|
|
|
Adjustment to prior year's provision versus statutory tax returns |
|
|
( |
) |
|
|
|
|
Other |
|
|
|
|
|
— |
|
|
Change in valuation allowance |
|
|
|
|
|
|
||
|
|
|
— |
|
|
|
— |
|
The Company has
The Company files U.S. federal, U.S. state, and Canadian income tax returns with varying statues of limitations. The tax years from remain open to examination due to the carryover of unused NOL carryforwards and tax credits. The Company currently is not under examination by any tax authority.
Internal Revenue Code (“IRC”) Section 382 and 383 places a limitation on the amount of taxable income that can be offset by NOL and credit carryforwards after a change in control (generally greater than
The CARES Act, was enacted March 27, 2020. Among the business provision, the CARES Act provided for various payroll tax incentives, changes to net operating loss carryback and carryforward rules, business interest expense limitation increases, and bonus depreciation on qualified improvement property. Additionally, the Consolidated Appropriations Act of 2021 was signed on December 27, 2020, which provided additional COVID-19 relief provisions for businesses. The Company has evaluated the impact of both the Acts and has determined that any impact is not material to its financial statements.
88
The Company has the following obligations over the next five fiscal years ending June 30, 2027:
Clinical development
The Company has entered into contracts for drug manufacturing, clinical study management and safety related to its clinical trials for a total of $
Office lease
The Company currently rents its shared head office on a
|
|
Year ended |
|
|
Year ended |
|
||
Series B Preferred Stock common stock dividend (note 8) |
|
|
— |
|
|
|
|
|
Series C Preferred Stock common stock dividend (note 8) |
|
|
|
|
|
— |
|
|
Deemed dividend recognized on beneficial conversion features of Series C Preferred stock issuance (note 8) |
|
|
— |
|
|
|
|
|
Non-cash issue costs (note 8) |
|
|
|
|
|
|
||
Issue costs in accounts payable |
|
|
|
|
|
— |
|
|
Cashless exercise of Series C warrants (note 8) |
|
|
— |
|
|
|
|
|
Conversion of Series B Preferred Stock to common stock (note 8) |
|
|
— |
|
|
|
|
|
Conversion of Series C Preferred Stock to common stock (note 8) |
|
|
|
|
|
|
||
Income taxes paid |
|
|
— |
|
|
|
— |
|
Interest paid |
|
|
— |
|
|
|
— |
|
Market risk
Market risk is the risk that changes in market prices, such as foreign exchange rates, interest rates and equity prices will affect the Company’s income or valuation of its financial instruments.
The Company is exposed to financial risk related to fluctuation of foreign exchange rates. Foreign currency risk is limited to the portion of the Company’s business transactions denominated in currencies other than the United Sates dollar, primarily general and administrative expenses incurred in Canadian dollars. The Company believes that the results of operations, financial position and cash flows would be affected by a sudden change in foreign exchange rates but would not impair or enhance its ability to pay its Canadian dollar accounts payable. The Company manages foreign exchange risk by converting its US$ to CA$ as needed. The Company maintains the majority of its cash in US$. As of June 30, 2022, Canadian dollar denominated accounts payable and accrued liabilities exposure in US$ totaled $
a) Foreign exchange risk
89
Balances in foreign currencies at June 30, 2022 and 2021 were as follows:
|
|
June 30, |
|
|
June 30, |
|
||
Trade payables |
|
|
|
|
|
|
||
Cash |
|
|
|
|
|
|
||
Interest, taxes, and other receivables |
|
|
|
|
|
|
||
b) Interest rate risk
The Company is subject to interest rate risk on its cash and cash equivalents and believes that the results of operations, financial position and cash flows would not be significantly affected by a sudden change in market interest rates relative to the investment interest rates due to the short-term nature of the investments. As of June 30, 2022, cash and cash equivalents held by the Company were $
The only financial instruments that expose the Company to interest rate risk are its cash and cash equivalents.
Liquidity risk
Liquidity risk is the risk that the Company will encounter difficulty in raising funds to meet cash flow requirements associated with financial instruments. The Company continues to manage its liquidity risk based on the outflows experienced for the period ended June 30, 2022, and is undertaking efforts to conserve cash resources wherever possible. The maximum exposure of the Company’s liquidity risk is $
Credit risk
Credit risk arises from cash and cash equivalents, deposits with banks, financial institutions, and contractors as well as outstanding receivables. The Company limits its exposure to credit risk, with respect to cash and cash equivalents, by placing them with high quality credit financial institutions. The Company’s cash equivalents consist primarily of operating funds with commercial banks. Of the amounts on deposit with financial institutions, the following table summarizes the amounts at risk should the financial institutions with which the deposits are held cease trading:
The maximum exposure of the Company’s credit risk is $
|
|
Cash and |
|
|
Insured |
|
|
Non- |
|
|||
|
|
|
|
|
|
|
|
|
|
|||
Concentration of credit risk
Financial instruments that subject the Company to credit risk consist primarily of cash and cash equivalents.
The Company places its cash and cash equivalents in accredited financial institutions and therefore the Company’s management believes these funds are subject to minimal credit risk. The Company has no significant off-balance sheet concentrations of credit risk such as foreign currency exchange contracts, option contracts or other hedging arrangements.
The Company has evaluated its subsequent events from June 30, 2022, through the date these consolidated financial statements were issued and has determined that there are no subsequent events requiring disclosure in these consolidated financial statements other than the items noted below.
Equity Purchase Agreement
On August 2, 2022, the Company entered into a purchase agreement, dated as of August 2, 2022 (the “Purchase Agreement”) with Lincoln Park Capital Fund, LLC (“Lincoln Park”), pursuant to which Lincoln Park has committed to purchase up to $
90
The Company issued
Pursuant to the Purchase Agreement, the Company has the right, in its sole discretion, to present Lincoln Park with a purchase notice directing Lincoln Park to purchase up to
If the Company directs Lincoln Park to purchase the maximum number of shares of common stock that the Company may sell in a Regular Purchase, then in addition to such Regular Purchase, and subject to certain conditions and limitations in the Purchase Agreement, the Company may direct Lincoln Park to purchase additional shares of common stock in an “accelerated purchase” (each, an “Accelerated Purchase”) and an “additional accelerated purchase” (each, an “Additional Accelerated Purchase”) (including multiple Additional Accelerated Purchases on the same trading day) as provided in the Purchase Agreement. The purchase price per share for each Accelerated Purchase and Additional Accelerated Purchase will be based on market prices of the common stock on the applicable purchase date for such Accelerated Purchases and such Additional Accelerated Purchases.
The aggregate number of shares that the Company can sell to Lincoln Park under the Purchase Agreement may in no case exceed
The Purchase Agreement may be terminated by the Company at any time, at its sole discretion, without any cost or penalty, by giving one business day notice to Lincoln Park to terminate the Purchase Agreement.
Subsequent to June 30, 2022, the Company sold
Series C Preferred Stock
On August 19, 2022, the Company paid the common stock dividend on its Series C Preferred Stock as well as the Series C Agent Warrants. The common stock dividend corresponds to the
Stock Options
Subsequent to June 30, 2022,
Restricted Stock Units
Subsequent to June 30, 2022,
Clinical Trials Deposit
Subsequent to June 30, 2022, the Company paid an additional $
91
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures are controls and other procedures that are designed to ensure that information required to be disclosed in our reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in our reports filed under the Exchange Act is accumulated and communicated to management, including our principal executive officer and our principal financial officer, as appropriate, to allow timely decisions regarding required disclosure. As of the end of the period covered by this report, we carried out an evaluation under the supervision and with the participation of our management, including our Chief Executive Officer (CEO) and our Chief Financial Officer (CFO), of the effectiveness of the design and operation of our disclosure controls and procedures in ensuring that material information required to be disclosed in our reports filed or submitted under the Exchange Act, has been made or known to them in a timely fashion. Based on this evaluation, our CEO and CFO concluded that the Company’s disclosure controls and procedures were effective as of June 30, 2022.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our management assessed, with the oversight of the board of directors, the effectiveness of our internal control over financial reporting as of June 30, 2022. In making this assessment, management used the criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based upon the evaluation, our management concluded that our internal control over financial reporting was effective as of June 30, 2022.
Changes in Control Over Financial Reporting
There have been no changes in our internal control over financial reporting identified in management’s evaluation pursuant to Rules 13a-15(d) or 15d-15(d) of the Exchange Act during the quarter ended June 30, 2022 that materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Limitations on Effectiveness of Controls and Procedures
In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable, not absolute, assurance of achieving the desired control objectives. Because of the inherent limitations in internal control over financial reporting, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision making can be faulty and that breakdowns can occur because of a simple error or mistake. Controls can also be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls is based in part on certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with policies or procedures. In addition, the design of disclosure controls and procedures must reflect the fact that there are resource constraints and that management is required to apply judgment in evaluating the benefits of possible controls and procedures relative to their costs. Because of the inherent limitations in a cost-effective controlled system, misstatements due to error or fraud may occur and not be detected.
Item 9B. Other Information.
On July 1, 2022, a total of 300,000 stock options were issued to the Company’s three independent directors. The stock options are exercisable at $0.255 per share and vest in 12 equal monthly installments beginning on August 1, 2022. All of the stock options have a 10-year term and are subject to cancellation upon the grantees’ termination of service for the Company, with certain exceptions.
On August 1, 2022, a total of 2,641,154 stock options were issued to officers of the Company. The stock options are exercisable at $0.1757 per share and vest as to 25% on August 1, 2023, with the remainder in 36 equal monthly installments beginning on September 1, 2023. All of the stock options have a 10-year term and are subject to cancellation upon the grantees’ termination of service for the Company, with certain exceptions. In addition, on August 1, 2022, a total of 880,319 restricted stock units ("RSU") were issued to officers of the Company. The RSU vest in four equal annual installments beginning on August 1, 2023.
Item 9C. Disclosure Regarding Foreign Jurisdictions That Prevent Inspections.
Not applicable.
92
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
Below are the names and certain information regarding our executive officers and directors.
Name |
|
Age |
|
Position |
Robert E. Hoffman |
|
56 |
|
President, CEO, Director, and Chairman of the Board |
Dennis Brown, PhD |
|
73 |
|
Chief Scientific Officer |
Scott Praill |
|
56 |
|
Chief Financial Officer |
Robert J. Toth, Jr. |
|
59 |
|
Director |
Laura Johnson |
|
58 |
|
Director |
Tamara A. Seymour |
|
64 |
|
Director |
Robert E. Hoffman has served as our President and Chief Executive Officer since November 8, 2021, as our Chairman since June 2, 2018, and as a director since April 11, 2018. He has served as a member of Aslan Pharmaceuticals, Inc.’s (NASDAQ:ASLN) board of directors since October 2018, and as a member of Antibe Therapeutics Inc.’s (TSE:ATE) board of directors since November 2020. Mr. Hoffman served as Senior Vice President and Chief Financial Officer of Heron Therapeutics, Inc. (NASDAQ:HRTX) a publicly-held pharmaceutical company from April 2017 to October 2020. Prior to joining Heron Therapeutics, Inc., Mr. Hoffman served as Executive Vice President and Chief Financial Officer of Innovus Pharmaceuticals, Inc. (OTC:INNV), a publicly-held pharmaceutical company, from September 2016 to April 2017. From July 2015 to September 2016, Mr. Hoffman served as Chief Financial Officer of AnaptysBio, Inc. (NASDAQ:ANAB), a publicly-held biotechnology company. From June 2012 to July 2015, Mr. Hoffman served as the Senior Vice President, Finance and Chief Financial Officer of Arena Pharmaceuticals, Inc. (NASDAQ:ARNA), or Arena, a publicly-held biopharmaceutical company. From August 2011 to June 2012 and previously from December 2005 to March 2011, he served as Arena’s Vice President, Finance and Chief Financial Officer and in a number of various roles of increasing responsibility from 1997 to December 2005. From March 2011 to August 2011, Mr. Hoffman served as Chief Financial Officer for Polaris Group, a biopharmaceutical drug company. Mr. Hoffman formerly served as a member of the board of directors of Kura Oncology, Inc. (NASDAQ:KURA), a cancer research company, CombiMatrix Corporation, a molecular diagnostics company, MabVax Therapeutics Holdings, Inc., a biopharmaceutical company and Aravive, Inc., a clinical stage biotechnology company. Mr. Hoffman serves as a member of the steering committee of the Association of Bioscience Financial Officers. Mr. Hoffman formerly served as a director and President of the San Diego Chapter of Financial Executives International and was an advisor to the Financial Accounting Standard Board (FASB) for 10 years (2010 to 2020) advising the United States accounting rulemaking organization on emerging issues and new financial guidance. Mr. Hoffman holds a B.B.A. from St. Bonaventure University, and is licensed as a C.P.A. (inactive) in the State of California. Mr. Hoffman’s financial and executive business experience qualifies him to serve on our board of directors.
Dennis Brown, PhD, has served as Kintara’s chief scientific officer since January 25, 2013. He also served as a director of the Company from February 11, 2013 to April 11, 2018. Dr. Brown is one of our founders and has served as Chief Scientific Officer and director of Del Mar (BC) since inception. Dr. Brown has more than thirty years of drug discovery and development experience. He has served as Chairman of Mountain View Pharmaceutical’s board of directors since 2000 and is the President of Valent. In addition, since March 2020 he has served as a director of Rakovina Therapeutics, Inc., a Canadian public company (TSXV:RKV). In 1999 he founded ChemGenex Therapeutics, which merged with a publicly traded Australian company in 2004 to become ChemGenex Pharmaceuticals (ASX: CXS/NASDAQ: CXSP), of which he served as President and a Director until 2009. He was previously a co-founder of Matrix Pharmaceutical, Inc., where he served as Vice President (VP) of Scientific Affairs from 1985-1995 and as VP, Discovery Research, from 1995-1999. He also previously served as an Assistant Professor of Radiology at Harvard University Medical School and as a Research Associate in Radiology at Stanford University Medical School. He received his B.A. in Biology and Chemistry (1971), M.S. in Cell Biology (1975) and Ph.D. in Radiation and Cancer Biology (1979), all from New York University. Dr. Brown is an inventor of many issued U.S. patents and applications, many with foreign counterparts.
Scott Praill has served as our chief financial officer since January 29, 2013, and previously served as a consultant to Del Mar (BC). From 2004 to 2012 Mr. Praill was an independent consultant providing accounting and administrative services to companies in the resource industry. From November 1999 to October 2003 Mr. Praill was Director of Finance at Inflazyme Pharmaceuticals Inc. Mr. Praill completed his articling at Price Waterhouse (now PricewaterhouseCoopers LLP) and obtained his Chartered Professional Accountant designation in 1996. Mr. Praill obtained his Certified Public Accountant (Illinois) designation in 2001. Mr. Praill received a Financial Management Diploma (Honors), from British Columbia Institute of Technology in 1993, and a Bachelor of Science from Simon Fraser University in 1989.
Robert J. Toth, Jr. has served as our director since August 20, 2013, and serves as Chair of our Compensation Committee. Since 2005, Mr. Toth has primarily been managing his personal investment portfolio. From 2004-2005, Mr. Toth served as a consulting analyst to Narragansett Asset Management, a New York-based healthcare-focused hedge fund, where he advised the firm on biotechnology investments. From 2001-2003, he was the Senior Portfolio Manager for San Francisco-based EGM Capital’s
93
Medical Technology hedge fund, where he was responsible for managing and maintaining a dedicated medical technology portfolio. Mr. Toth began his Wall Street career in 1996 as an Equity Research Associate for Vector Securities International, a healthcare-focused brokerage firm. From 1997-1999 he served as Senior Biotechnology Analyst. He joined Prudential Securities as Senior Vice President and Biotechnology Analyst where he served from 1999-2001 following Prudential’s acquisition of Vector. His responsibilities included the analysis of commercial, clinical and scientific fundamentals of oncology and genomics-based biotechnology companies on behalf of institutional investors. Mr. Toth was named to the Wall Street Journal’s Allstar List for stock picking in 1999. Mr. Toth received an MBA from the University of Washington and Bachelor of Science degrees in Biological Sciences and Biochemistry from California Polytechnic State University, San Luis Obispo. Mr. Toth’s financial and biotechnology industry knowledge and experience quality him to serve on our board of directors.
Laura Johnson has served as our director since June 26, 2020 and serves as Chair of our Nominating and Corporate Governance Committee. Ms. Johnson currently serves as the President and Chief Executive Officer of Next Generation Clinical Research, a contract research organization that Ms. Johnson founded in 1999. Additionally, Ms. Johnson is the President and Chief Executive Officer of Eufaeria Biosciences, Inc., a development biotechnology company that she founded in 2016. Ms. Johnson is also a founder and former member of the board of directors of SB Bancorp, Inc., a financial holding company, and Settlers Bank, Inc., a Wisconsin chartered business bank. In addition, Ms. Johnson serves as a member of the board of directors of La Jolla Pharmaceutical Company (Nasdaq: LJPC), a biopharmaceutical company, since 2013, Harmony Hill Farm Sanctuary since 2019 and Agrace HospiceCare from 2013 to 2016. In 2008 and 2010, she was honored as a biotechnology entrepreneur by the national organization, Women in Bio, and in 2008 received the Rising Star Award by the Wisconsin Biotech and Medical Device Association. Most recently, she was the recipient of the Wisconsin Biohealth Business Award at the BioForward Annual Biohealth Summit in October 2019. Ms. Johnson holds a nursing degree from The University of the State of New York-Albany. Ms. Johnson’s biotechnology industry and executive knowledge and experience qualify her to serve on our board of directors.
Tamara A. Seymour has served as our director since April 29, 2021, and Chair of our Audit Committee since July 1, 2021. Ms. Seymour has more than 30 years of life sciences industry experience including 20 years as a chief financial officer. She currently serves as a board member and audit committee chair of Artelo Biosciences, Inc. (Nasdaq: ARTL), and KemPharm, Inc. (Nasdaq: KMPH), both publicly-traded clinical-development stage companies. Ms. Seymour served on the board of directors of Beacon Discovery, Inc. from 2018 until their acquisition in 2021. Ms. Seymour was Interim Chief Financial Officer of Immunic, Inc. (Nasdaq: IMUX), a publicly-traded clinical-stage drug development company in 2019. She served as Chief Financial Officer of Signal Genetics, Inc., (Nasdaq: VRDN) a publicly-traded molecular diagnostics company, from 2014 to 2017, HemaQuest Pharmaceuticals, Inc., a venture-backed clinical-stage drug development company, from 2010 to 2014 and Favrille, Inc., a previously publicly-traded clinical-stage drug development company, from 2001 to 2009. While at these companies, she led multiple private and public financings, including Favrille’s IPO. In addition, she was instrumental in M&A transactions and led the finance, investor relations, human resources, administration and managed care and payor reimbursement functions. Ms. Seymour is a Certified Public Accountant (inactive). She received an MBA, with an emphasis in Finance, from Georgia State University, and a bachelor's degree in Business Administration, with an emphasis in Accounting from Valdosta State University. Ms. Seymour also participated in an executive management program at Kellogg Graduate School of Management at Northwestern University. Ms. Seymour’s professional experience and financial expertise qualify her to serve on our board of directors.
Our chief executive and chief financial officers are full-time employees and devote 100% of their business time to us. Our consulting agreement with Dr. Brown does not specify the amount of time Dr. Brown is required to devote to us but does require that Dr. Brown provide us with the full benefit of his knowledge, expertise and ingenuity, and prohibits Dr. Brown from engaging in any business, enterprise or activity contrary to or that would detract from our business.
Our directors are appointed for a one-year term to hold office until the next annual general meeting of our stockholders or until removed from office in accordance with our bylaws and the provisions of the Nevada Revised Statutes.
Our officers are appointed by our board of directors and serve at its pleasure.
94
Involvement in Certain Legal Proceedings
We are not involved in any litigation that we believe could have a material adverse effect on our financial position or results of operations. There is no action, suit, proceeding, inquiry or investigation before or by any court, public board, government agency, self-regulatory organization or body pending or, to the knowledge of our executive officers, threatened against or affecting our Company or our officers or directors in their capacities as such.
Board Committees
The board of directors has formed an Audit Committee which currently consists of Tamara A. Seymour, Chair, Robert J. Toth, Jr., and Laura Johnson all of whom are independent (as that term is defined under the Nasdaq Marketplace Rules) and financially literate (as such qualification is interpreted by the board of directors in its business judgment). In addition, our board of directors has determined that Ms. Seymour qualifies as audit committee financial experts within the meaning of SEC regulations and The NASDAQ Marketplace Rules.
The board of directors has also formed a Nominating and Corporate Governance Committee which consists of Laura Johnson, Chair, Robert J. Toth, Jr., and Tamara A. Seymour. The Nominating and Corporate Governance Committee assists the board of directors in fulfilling its oversight responsibilities relating to corporate governance practices and policies.
In addition, the board of directors has formed a Compensation Committee which consists of Robert J. Toth, Jr., Chair, Tamara A. Seymour, and Laura Johnson. The Compensation Committee assists the board of directors in fulfilling its oversight responsibilities relating to compensation matters, including compensation of the directors and our senior management and the administration of our compensation plans.
Each of our Audit Committee, Nominating and Corporate Governance Committee, and Compensation Committee operates pursuant to a charter that is posted under the “Investors” tab under Corporate Governance on our website, which is located at www.kintara.com.
Nomination of Directors
The Nominating and Corporate Governance Committee of the board of directors assesses potential candidates to fill perceived needs on the board of directors for required skills, expertise, independence and other factors. The Nominating and Corporate Governance Committee consists of independent directors only.
Orientation and Continuing Education
New members of the board of directors are provided with sufficient information to ensure that they are familiarized with us, our policies, and the mandates of the board of directors. Members of the board of directors are encouraged to communicate with management, legal counsel and, where applicable, our auditors and technical consultants to keep themselves current with industry developments and applicable legal, accounting and regulatory changes.
Board Leadership Structure and Role in Risk Oversight
Robert E. Hoffman serves as our Chief Executive Officer, President, and chairman of our board of directors. We have not adopted a formal policy on whether the Chief Executive Officer and Chairman positions should be separated.
Our board of directors is primarily responsible for overseeing our risk management processes. The board of directors receives and reviews periodic reports from management, auditors, legal counsel, and others, as considered appropriate regarding our assessment of risks. The board of directors focuses on the most significant risks facing us and our general risk management strategy, and also ensures that risks undertaken by us are consistent with the board’s appetite for risk. While the board of directors oversees our risk management, management is responsible for the day-to-day risk management processes. We believe this division of responsibilities is the most effective approach for addressing the risks we face and that our board leadership structure supports this approach.
Code of Ethics
We have adopted a Code of Ethics and Business Conduct that applies to all of our executive officers, financial and accounting officers, our directors, our financial managers and all of our employees. Our board of directors is committed to a high standard of corporate governance practices and, through its oversight role, encourages and promotes a culture of ethical business conduct. A copy of our Code of Ethics and Business Conduct is posted under the “Investors” tab under Corporate Governance on our website, which is located at www.kintara.com.
95
Assessments
The board of directors assesses, on an ongoing basis, its overall performance and that of its committees in order to determine whether they are performing effectively. The board of directors also assesses, on an ongoing basis, the effectiveness and contribution of each of our directors, having regard to the competencies and skills each director is expected to bring to the board of directors.
Item 11. Executive Compensation.
Our board of directors has formed a Compensation Committee. The Compensation Committee is responsible for reviewing and approving management compensation, including salaries, bonuses, and equity compensation. We seek to provide competitive compensation arrangements that attract and retain key talent necessary to achieve our business objectives. At our 2021 annual meeting of stockholders, stockholders voted, on an advisory, non-binding basis, to approve the compensation paid to the company’s named executive officers, as disclosed in the proxy statement for the 2021 annual meeting. At our 2018 annual meeting, our stockholders voted, on an advisory, non-binding basis, that such votes on named executive officer compensation should be held every three years. The next advisory, non-binding vote to approve named executive officer compensation is expected to occur in connection with the 2024 annual meeting of stockholders.
Summary Compensation Table
The following table presents information regarding the total compensation awarded to, earned by, or paid to our Chief Executive Officer and the two most highly-compensated executive officers (other than the Chief Executive Officer) who were serving as executive officers as of June 30, 2022 and June 30, 2021 for services rendered in all capacities to us for the years ended June 30, 2022 and June 30, 2021. These individuals are our Named Executive Officers for 2022.
Name and Principal Position |
|
Period |
|
Salary |
|
|
Bonus |
|
|
Equity |
|
|
Total |
|
||||
Robert E. Hoffman, President and CEO(1) |
|
Year Ended |
|
|
356,619 |
|
|
|
181,811 |
|
|
|
2,622,597 |
|
|
|
3,161,028 |
|
|
|
Year Ended |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
Saiid Zarrabian, Former President and CEO and Former Head of Strategic Partnerships(2) |
|
Year Ended |
|
|
563,297 |
|
|
|
119,530 |
|
|
|
— |
|
|
|
682,827 |
|
|
|
Year Ended |
|
|
520,000 |
|
|
|
180,830 |
|
|
|
4,270,860 |
|
|
|
4,971,690 |
|
Scott Praill, Chief Financial Officer(3) |
|
Year Ended |
|
|
312,000 |
|
|
|
127,920 |
|
|
|
— |
|
|
|
439,920 |
|
|
|
Year Ended |
|
|
290,000 |
|
|
|
79,808 |
|
|
|
954,150 |
|
|
|
1,323,958 |
|
Dennis Brown, Chief Scientific Officer(4) |
|
Year Ended |
|
|
206,000 |
|
|
|
41,277 |
|
|
|
— |
|
|
|
247,277 |
|
|
|
Year Ended |
|
|
200,000 |
|
|
|
33,544 |
|
|
|
532,149 |
|
|
|
765,693 |
|
John Liatos, Former Senior V.P., Business Development(5) |
|
Year Ended |
|
|
334,769 |
|
|
|
— |
|
|
|
— |
|
|
|
334,769 |
|
|
|
Year Ended |
|
|
340,000 |
|
|
|
105,725 |
|
|
|
796,986 |
|
|
|
1,242,711 |
|
96
On November 8, 2021, Mr. Hoffman was granted 3,519,170 stock options that are exercisable at $0.96 per share until November 8, 2031. On September 22, 2021, while still an independent director, Mr. Hoffman was granted 100,000 stock options that are exercisable at $1.24 per share until September 22, 2031.Mr. Hoffman's bonus for the fiscal year ended June 30, 2022, was $181,811.
On November 8, 2021, in connection with the appointment of Mr. Hoffman, the board and Mr. Zarrabian agreed to enter into a Second Amended Employment Agreement pursuant to which Mr. Zarrabian was appointed Head of Strategic Partnerships. Pursuant to the terms of the agreement, Mr. Zarrabian was entitled to receive an annual base salary of $285,000 and was eligible to receive a fiscal year target bonus of up to 40% of base salary, subject to the achievement of performance targets or criteria established by the board for such year. The employment agreement was terminable by us with or without cause (as defined therein). In the event we terminated Mr. Zarrabian’s employment without cause, or if Mr. Zarrabian resigned for good reason (as defined in the employment agreement), we would have been required to pay Mr. Zarrabian continued payment of his base salary for nine months, any earned, but unpaid bonus for the preceding year, and a prorated bonus for the year of termination based on performance through the date of termination. In addition, in that event Mr. Zarrabian would have been provided an additional six months of vesting credit for any outstanding stock options, and continued health coverage during the severance period. In the event that a termination of Mr. Zarrabian’s employment under such circumstances occurred during a period beginning 60 days before a definitive corporate transaction agreement was entered into that would result in a change in control (as defined in the employment agreement), or within 12 months following a change in control, the foregoing separation pay and benefits would have applied except that his base salary would continue for 12 months (instead of nine months), he would have received 100% of his target bonus (rather than a prorated bonus), and his stock options would have been fully vested. In connection with Mr. Zarrabian’s appointment as Head of Strategic Partnerships, the vesting terms of certain of Mr. Zarrabian’s outstanding unvested stock option were amended, such that an aggregate of 754,175 shares subject to such stock option that were unvested and subject to continued service requirements were then subject to vesting based on both Mr. Zarrabian’s continued service and achievement of certain milestones established by the board of directors.
On November 11, 2020, 279,675 stock options previously granted to Mr. Zarrabian at $0.61 per share had their vesting accelerated such that the 279,675 stock options all vested on November 11, 2020. Mr. Zarrabian’s bonus for the fiscal year ended June 30, 2021 was $180,830.
On May 20, 2022, we mutually agreed with Mr. Zarrabian that he would step down from his role as Head of Strategic Partnerships and as a member of the Board, effective as of May 23, 2022. In connection with Mr. Zarrabian’s separation on May 20, 2022, we entered into a separation and general release agreement with Mr. Zarrabian which provides, among other things, for Mr. Zarrabian to receive continued payments of nine months of his annual base salary, equal to the sum of $213,750, commencing on the first regular payroll date that is after the separation date and paid in installments in accordance with our regular payroll practices, a one-time bonus payment of $24,826.67 in connection with his service as our Head of Strategic Partnerships, reimbursement of healthcare coverage payments for a period of up to nine months following the separation date, continued payments of life insurance premiums for a period of up to nine months following the separation date, and an additional six months of service vesting credit for each of his stock options outstanding as of the separation date, and all of his vested stock options, including any options so accelerated, remaining exercisable for up to a nine-month period measured from the separation date (or earlier expiration of the options term). The separation agreement further provides for general release and non-disparagement provisions in our favor. In
97
addition, Mr. Zarrabian will be subject to non-solicitation provisions, which will apply for a period of twelve months following the separation date.
On September 15, 2020, he was granted 606,557 stock options that are exercisable at $1.70 per share until September 15, 2030.
On September 15, 2020, Dr. Brown was granted 89,437 stock options that are exercisable at $1.70 per share until September 15, 2030, and on September 22, 2020, he was granted 60,000 stock options that are exercisable at $1.36 per share until September 22, 2030.
On September 15, 2020 Mr. Liatos was granted 506,647 stock options that are exercisable at $1.70 per share until September 15, 2030. Mr. Liatos’ performance bonus for the fiscal year ended June 30, 2021 was $49,725 and he also received a retention bonus of $56,000.
On October 25, 2021, we mutually agreed with Mr. Liatos that Mr. Liatos would step down from his role as senior vice president, business development. In connection with the termination of Mr. Liatos’s services, on October 29, 2021, we entered into a separation and general release agreement with Mr. Liatos. In substantial part, the terms of the severance payable under the separation agreement will be governed by the terms of Mr. Liatos’s amended and restated employment agreement, dated March 1, 2018. As contemplated in that agreement, among other things, Mr. Liatos will receive continued payments of eight months of his annual base salary, equal to the sum of $226,667, commencing on the first regular payroll date that is at least 60 days after the termination date and paid in installments in accordance with our regular payroll practices, reimbursement of healthcare continuation payments under the Consolidated Omnibus Budget Reconciliation Act for a period of up to eight months following the termination date, and an additional six months of service vesting credit for each of his stock options outstanding as of the termination date, and all of his vested options, including any options so accelerated, remaining exercisable for up to a
98
twelve-month period measured from the termination date (or earlier expiration of the options term). In addition, Mr. Liatos will be subject to non-compete and non-solicitation provisions, which will apply for a period of twelve months following the termination date.
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth outstanding equity awards to our named executive officers as of June 30, 2022.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock awards |
|
|||||||||||
Name |
|
Number of |
|
|
Number of |
|
|
Equity |
|
|
Option |
|
|
Option |
|
Equity |
|
|
Equity |
|
||||||
Robert E. Hoffman |
|
|
3,600 |
|
|
|
— |
|
|
|
— |
|
|
|
10.60 |
|
|
April 13, 2028 |
|
|
— |
|
|
|
— |
|
|
|
|
4,000 |
|
|
|
— |
|
|
|
— |
|
|
|
6.10 |
|
|
November 8, 2028 |
|
|
— |
|
|
|
— |
|
|
|
|
75,000 |
|
|
|
— |
|
|
|
— |
|
|
|
0.61 |
|
|
September 5, 2029 |
|
|
— |
|
|
|
— |
|
|
|
|
120,000 |
|
|
|
— |
|
|
|
— |
|
|
|
1.70 |
|
|
September 15, 2030 |
|
|
— |
|
|
|
— |
|
|
|
|
75,000 |
|
(1) |
|
25,000 |
|
|
|
— |
|
|
|
1.24 |
|
|
September 22, 2031 |
|
|
— |
|
|
|
— |
|
|
|
|
— |
|
(2) |
|
3,519,170 |
|
|
|
— |
|
|
|
0.96 |
|
|
November 8, 2031 |
|
|
— |
|
|
|
— |
|
Saiid Zarrabian |
|
|
3,600 |
|
|
|
— |
|
|
|
— |
|
|
|
21.10 |
|
|
February 23, 2023 |
|
|
— |
|
|
|
— |
|
|
|
|
12,000 |
|
|
|
— |
|
|
|
— |
|
|
|
8.70 |
|
|
February 23, 2023 |
|
|
— |
|
|
|
— |
|
|
|
|
83,647 |
|
|
|
— |
|
|
|
— |
|
|
|
9.83 |
|
|
February 23, 2023 |
|
|
— |
|
|
|
— |
|
|
|
|
457,650 |
|
|
|
— |
|
|
|
— |
|
|
|
0.61 |
|
|
February 23, 2023 |
|
|
— |
|
|
|
— |
|
|
|
|
1,960,816 |
|
|
|
— |
|
|
|
— |
|
|
|
1.70 |
|
|
February 23, 2023 |
|
|
— |
|
|
|
— |
|
Scott Praill |
|
|
8,750 |
|
|
|
— |
|
|
|
— |
|
|
|
42.00 |
|
|
August 15, 2023 |
|
|
— |
|
|
|
— |
|
|
|
|
3,740 |
|
|
|
— |
|
|
|
— |
|
|
|
49.50 |
|
|
February 17, 2027 |
|
|
— |
|
|
|
— |
|
|
|
|
10,000 |
|
|
|
— |
|
|
|
— |
|
|
|
6.10 |
|
|
November 8, 2028 |
|
|
— |
|
|
|
— |
|
|
|
|
99,331 |
|
(3) |
|
9,035 |
|
|
|
— |
|
|
|
0.61 |
|
|
September 5, 2029 |
|
|
— |
|
|
|
— |
|
|
|
|
353,808 |
|
(4) |
|
252,749 |
|
|
|
— |
|
|
|
1.70 |
|
|
September 15, 2030 |
|
|
— |
|
|
|
— |
|
Dennis Brown |
|
|
8,750 |
|
|
|
— |
|
|
|
— |
|
|
|
42.00 |
|
|
August 15, 2023 |
|
|
— |
|
|
|
— |
|
|
|
|
9,360 |
|
|
|
— |
|
|
|
— |
|
|
|
49.50 |
|
|
February 17, 2027 |
|
|
— |
|
|
|
— |
|
|
|
|
91,673 |
|
(3) |
|
8,327 |
|
|
|
— |
|
|
|
0.61 |
|
|
September 5, 2029 |
|
|
— |
|
|
|
— |
|
|
|
|
145,824 |
|
(5) |
|
104,176 |
|
|
|
— |
|
|
|
0.74 |
|
|
November 12, 2029 |
|
|
— |
|
|
|
— |
|
|
|
|
50,988 |
|
(4) |
|
36,449 |
|
|
|
— |
|
|
|
1.70 |
|
|
September 15, 2030 |
|
|
— |
|
|
|
— |
|
|
|
|
34,990 |
|
(5) |
|
25,010 |
|
|
|
— |
|
|
|
1.36 |
|
|
September 22, 2030 |
|
|
— |
|
|
|
— |
|
John Liatos |
|
|
267,387 |
|
|
|
— |
|
|
|
— |
|
|
|
1.70 |
|
|
October 25, 2022 |
|
|
— |
|
|
|
— |
|
99
Director Compensation
Director compensation is intended to provide an appropriate level of remuneration considering the responsibilities, time requirements, and accountability of the directors.
The following table sets forth director compensation for the fiscal year ended June 30, 2022, paid by us (excluding compensation to our executive officers set forth in the summary compensation table above).
Name |
|
Fees |
|
|
Stock |
|
|
Option |
|
|
Non-Equity |
|
|
Nonqualified |
|
|
All Other |
|
|
Total ($) |
|
|||||||
Robert E. Hoffman(3) |
|
|
30,868 |
|
|
|
— |
|
|
|
89,963 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
120,831 |
|
Robert J, Toth, Jr. |
|
|
61,500 |
|
|
|
— |
|
|
|
89,963 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
151,463 |
|
Laura Johnson |
|
|
57,524 |
|
|
|
— |
|
|
|
89,963 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
147,487 |
|
Keith Murphy(4) |
|
|
29,167 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
29,167 |
|
Tamara A. Seymour |
|
|
59,855 |
|
|
|
— |
|
|
|
31,487 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
91,342 |
|
Risk Management
We do not believe risks arising from its compensation policies and practices for its employees are reasonably likely to have a material adverse effect on us.
100
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth certain information, as of September 26, 2022, with respect to the beneficial ownership of the outstanding common stock by (i) any holder of more than five (5%) percent; (ii) each of our named executive officers and directors; and (iii) our directors and executive officers as a group. Except as otherwise indicated, each of the stockholders listed below has sole voting and investment power over the shares beneficially owned.
Name of Beneficial Owner (1) |
|
Common |
|
Percentage of |
|
||||
Directors and Named Executive Officers |
|
|
|
|
|
|
|
||
Robert E. Hoffman |
|
|
1,237,393 |
|
(3) |
|
|
1.5 |
% |
Saiid Zarrabian |
|
|
2,573,803 |
|
(4) |
|
|
3.1 |
% |
John Liatos |
|
|
464,137 |
|
(5) |
|
* |
|
|
Scott Praill |
|
|
572,394 |
|
(6) |
|
* |
|
|
Dennis Brown, PhD |
|
|
443,594 |
|
(7) |
|
* |
|
|
Robert J. Toth, Jr. |
|
|
323,833 |
|
(8) |
|
* |
|
|
Laura Johnson |
|
|
239,667 |
|
(9) |
|
* |
|
|
Tamara A. Seymour |
|
|
82,917 |
|
(10) |
|
* |
|
|
All officers and directors as a group (6 persons) |
|
|
2,899,797 |
|
|
|
|
3.5 |
% |
* Less than 1%
101
Securities Authorized for Issuance Under Equity Compensation Plans
The following table sets forth the aggregate information of our equity compensation plans in effect as of June 30, 2022:
Plan (in thousands, except per share amounts) |
|
Number of |
|
|
Weighted- |
|
|
Number of |
|
|||
Equity compensation plans approved by security holders(1) |
|
|
8,693 |
|
|
$ |
1.33 |
|
|
|
12,996 |
|
Equity compensation plans not approved by security |
|
|
117 |
|
|
$ |
32.40 |
|
|
|
— |
|
Totals |
|
|
8,810 |
|
|
$ |
1.74 |
|
|
|
12,996 |
|
A total of 117 shares of our common stock, net of forfeitures, have been issued under the Legacy Plan and/or are subject to outstanding stock options granted under the Legacy Plan, and a total of 8,693 shares of our common stock have been issued under the 2017 Plan and/or are subject to outstanding stock options granted under the 2017 Plan leaving a potential 12,996 shares, net of exercises, of our common stock available for issuance under the Plan if all such options under the Legacy Plan were exercised and no new grants are made under the Legacy Plan. The maximum number of shares of our common stock with respect to which any one participant may be granted awards during any calendar year is 8% of our fully diluted shares of common stock on the date of grant (excluding the number of shares of our common stock issued under the 2017 Plan and/or the Legacy Plan or subject to outstanding awards granted under the 2017 Plan and/or the Legacy Plan). No award will be granted under the 2017 Plan on or after July 7, 2027, but awards granted prior to that date may extend beyond that date.
Certain Relationships and Related Transactions
Valent Technologies LLC
On September 12, 2010, Del Mar (BC) entered into a Patent Assignment Agreement (the “Assignment”) with Valent Technologies LLC pursuant to which Valent assigned to Del Mar (BC) its rights to patent applications and the prototype drug product related to VAL-083. In accordance with the Assignment the consideration paid by Del Mar (BC) was $250,000 to acquire the prototype drug product. In accordance with the terms of the Assignment, Valent is entitled to receive a future royalty on all revenues derived from the development and commercialization of VAL-083. In the event that Del Mar (BC) terminates the agreement, Del Mar (BC) may be entitled to receive royalties from Valent’s subsequent development of VAL-083 depending on the development milestones Del Mar (BC) has achieved prior to the termination of the Assignment. The Assignment has a term (on a country-by-country basis), of the later of ten years or until patent rights covered by the Assignment no longer exist, subject to earlier termination in the event Del Mar (BC) breaches its payment obligations and fails to remedy such breach within 60 days, or if either party materially beaches any of its obligations and does not cure such breach within 30 days after receipt of notice thereof.
Pursuant to a loan agreement dated February 3, 2011, between Del Mar (BC) and Valent, Valent loaned Del Mar $250,000 for the purchase of the prototype drug product under the Assignment. The loan is unsecured, bears interest at 3% per year, and is payable on demand. Effective September 30, 2014, we entered into and closed an agreement with Valent to exchange its loan, including
102
accrued interest to September 30, 2014, with Valent for 278,530 shares of our preferred stock. The preferred stock has an annual 3% dividend.
One of our officers, Dr. Dennis Brown, is a principal of Valent and as result Valent is a related party to us.
St. Cloud Investments, LLC
We acquired certain Miravant assets, including the REM-001 Therapy and the associated technology and intellectual property, through an asset purchase agreement with St. Cloud Investments, LLC (“St. Cloud”), dated November 26, 2012, as amended (the “St. Cloud Agreement”). St. Cloud was previously a Miravant creditor and acquired these Miravant assets pursuant to a foreclosure process St. Cloud completed under California law. Pursuant to the terms of the St. Cloud Agreement, we are obligated to make certain payments under the agreement. The amounts paid or owed under that agreement are as follows:
With respect to the $300,000 and $700,000 potential milestone payments referenced above (each a “Milestone Payment”), if either such Milestone Payment becomes payable, and in the event we elect to pay either such Milestone Payment in shares of our common stock, the value of the common stock will equal the price per share of the most recent financing, or, if we are considered to be a publicly-traded company, the average of the closing price per share of our common stock over the twenty (20) trading days following the first public announcement of the applicable event described above.
In addition, we must pay to St. Cloud and one of our former officers, Steven Rychnovsky, PhD, in the aggregate, a royalty fee of six percent (6%) of net sales during the royalty term on a country-by-country and product-by-product basis with St. Cloud receiving a royalty rate of four and eight tenths percent (4.8%) and Steven Rychnovsky, PhD, receiving a royalty of one and two tenths percent (1.2%). The royalty term for a product commences on the first commercial sale of the product, such as REM-001 Therapy, in any country, and the royalty fee must be paid within 30 days of each calendar quarter during which revenue is collected. The royalty term terminates on the later of (i) the invalidation, revocation, lapse or expiration of the last to expire valid claim on any patent acquired in the St. Cloud Agreement that would be infringed by the sale of the product in the country where the commercial sale takes place or (ii) the expiration of the period for which we hold exclusive marketing rights of the product in the country, if we were granted those rights under the St. Cloud Agreement.
Director Independence
Robert J. Toth, Jr., Laura Johnson, and Tamara A. Seymour are independent as that term is defined under the Nasdaq Marketplace Rules.
103
Item 14. Principal Accounting Fees and Services.
On July 31, 2019, Marcum LLP (“Marcum”), Certified Public Accountants, were appointed as our auditors.
The following is a summary of fees paid by us for professional services rendered by Marcum for the years ended June 30, 2022, and 2021.
|
|
Year Ended |
|
|
Year Ended |
|
||
Audit fees |
|
$ |
225,900 |
|
|
$ |
200,599 |
|
Audit related fees |
|
$ |
44,869 |
|
|
$ |
27,077 |
|
Tax fees |
|
$ |
— |
|
|
$ |
— |
|
All other fees |
|
$ |
— |
|
|
$ |
— |
|
Total fees |
|
$ |
270,769 |
|
|
$ |
227,676 |
|
Audit fees. Audit fees represent fees for professional services performed by Marcum for the audit of our annual financial statements and the review of our quarterly financial statements, as well as services that are normally provided in connection with statutory and regulatory filings or engagements.
Audit-related fees. Audit-related fees represent fees for assurance and related services performed by Marcum that are reasonably related to the performance of the audit or review of our financial statements.
Tax Fees. Marcum has not performed any tax compliance services for us during the years ended June 30, 2022, or 2021.
All other fees. Marcum has not received any other fees from us for the years ended June 30, 2022, or 2021.
In accordance with applicable laws, rules and regulations, our audit committee charter and pre-approval policies established by the audit committee require that the audit committee review in advance and pre-approve all audit and permitted non-audit fees for services provided to us by our independent registered public accounting firm. The services performed by, and the fees to be paid to, Marcum in 2022 and 2021, respectively, were approved by the audit committee.
104
PART IV
Item 15. Exhibits.
2.1 |
|
|
|
|
|
2.2†† |
|
|
|
|
|
3.1 |
|
|
|
|
|
3.2 |
|
|
|
|
|
3.3 |
|
|
|
|
|
3.4 |
|
|
|
|
|
3.5 |
|
|
|
|
|
3.6 |
|
|
|
|
|
3.7 |
|
|
|
|
|
3.8 |
|
|
|
|
|
3.9 |
|
|
|
|
|
3.10 |
|
|
|
|
|
3.11 |
|
|
|
|
|
3.12 |
|
|
|
|
|
3.13 |
|
|
|
|
|
3.14 |
|
|
|
|
|
3.15 |
|
|
|
|
|
3.16 |
|
|
|
|
|
3.17 |
|
|
|
|
|
105
3.18 |
|
|
|
|
|
4.1 |
|
|
|
|
|
4.2 |
|
|
|
|
|
4.3 |
|
|
|
|
|
4.4 |
|
|
|
|
|
4.5 |
|
|
|
|
|
4.6 |
|
|
|
|
|
4.7 |
|
|
|
|
|
4.8 |
|
|
|
|
|
4.9 |
|
|
|
|
|
4.10 |
|
|
|
|
|
4.11 |
|
|
|
|
|
4.12 |
|
|
|
|
|
10.1 |
|
|
|
|
|
10.2 |
|
|
|
|
|
10.3 |
|
|
|
|
|
10.4 |
|
|
|
|
|
10.5 |
|
|
|
|
|
10.6† |
|
|
|
|
|
10.7† |
|
|
|
|
|
106
10.8 |
|
|
|
|
|
10.9 |
|
|
|
|
|
10.10 |
|
|
|
|
|
10.11 |
|
|
|
|
|
10.12 |
|
|
|
|
|
10.13 |
|
|
|
|
|
10.14 |
|
|
|
|
|
10.15 |
|
|
|
|
|
10.16 |
|
|
|
|
|
10.17 |
|
|
|
|
|
10.18 |
|
|
|
|
|
10.19 |
|
|
|
|
|
10.20 |
|
|
|
|
|
10.21 |
|
|
|
|
|
10.22 |
|
|
|
|
|
10.23 |
|
|
|
|
|
10.24 |
|
|
|
|
|
10.25 |
|
|
|
|
|
107
10.26 |
|
|
|
|
|
10.27 |
|
|
|
|
|
10.28 |
|
|
|
|
|
10.29 |
|
|
|
|
|
10.30 |
|
|
|
|
|
10.31 |
|
|
|
|
|
10.32 |
|
|
|
|
|
10.33 |
|
Amendment to the 2017 Omnibus Equity Incentive Plan of Kintara Therapeutics, Inc.* |
|
|
|
10.34 |
|
|
|
|
|
10.35 |
|
|
|
|
|
21.1 |
|
|
|
|
|
23.1 |
|
|
|
|
|
31.1 |
|
|
|
|
|
31.2 |
|
|
|
|
|
32.1 |
|
|
|
|
|
32.2 |
|
|
|
|
|
EX-101.INS |
|
Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because XBRL tags are embedded within the Inline XBRL document. |
|
|
|
EX-101.SCH |
|
Inline XBRL Taxonomy Extension Schema Document |
|
|
|
EX-101.CAL |
|
Inline XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
EX-101.DEF |
|
Inline XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
EX-101.LAB |
|
Inline XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
EX-101.PRE |
|
Inline XBRL Taxonomy Extension Presentation Linkbase Document |
|
|
|
104 Cover Page Interactive Data File (embedded within the Inline XBRL document) |
||
† Confidential treatment is requested for certain confidential portions of this exhibit pursuant to Rule 24b-2 under the Exchange Act. In accordance with Rule 24b-2, these confidential portions have been omitted from this exhibit and filed separately with the Commission.
108
†† Schedule has been omitted pursuant to Item 601(a)(5) of Regulation S-K. The Company hereby undertakes to furnish copies of any of the omitted schedules upon request by the Securities and Exchange Commission.
* Filed herewith
** Furnished herewith
Item 16. Form 10-K Summary.
None.
109
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
KINTARA THERAPEUTICS, INC. |
||
|
|
|
|
Dated: September 27, 2022 |
By: |
/s/ Robert E. Hoffman |
|
|
|
Name: |
Robert E. Hoffman |
|
|
Title: |
Chief Executive Officer |
Dated: September 27, 2022 |
By: |
/s/ Scott Praill |
|
|
|
Name: |
Scott Praill |
|
|
Title: |
Chief Financial Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
SIGNATURE |
|
TITLE |
|
DATE |
|
|
|
|
|
/s/ Robert E. Hoffman |
|
Chief Executive Officer, Director |
|
September 27, 2022 |
Robert E. Hoffman |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
/s/ Scott Praill |
|
Chief Financial Officer |
|
September 27, 2022 |
Scott Praill |
|
(Principal Financial and Accounting Officer) |
|
|
|
|
|
|
|
/s/ Dennis Brown |
|
Chief Scientific Officer |
|
September 27, 2022 |
Dennis Brown |
|
|
|
|
|
|
|
|
|
/s/ Tamara A. Seymour |
|
Director |
|
September 27, 2022 |
Tamara A. Seymour |
|
|
|
|
|
|
|
|
|
/s/ Robert J. Toth |
|
Director |
|
September 27, 2022 |
Robert J. Toth |
|
|
|
|
|
|
|
|
|
/s/ Laura Johnson |
|
Director |
|
September 27, 2022 |
Laura Johnson |
|
|
|
|
110